Issue Ebook
cover story

Latest Issue
Just Accepted Online First List of Issues Document Types
Issue 11, 2025
RAPID COMMUNICATION
2025, 43(11): 1939-1949. DOI: 10.1007/s10118-025-3430-7Published(online): 2025-10-30Abstract Full text L-PDFAbstract

REVIEW
2025, 43(11): 1950-1972. DOI: 10.1007/s10118-025-3398-3Published(online): 2025-10-30Abstract Full text L-PDFAbstract
RESEARCH ARTICLE
2025, 43(11): 1973-1980. DOI: 10.1007/s10118-025-3431-6Published(online): 2025-10-30Abstract Full text L-PDFAbstract![Photocontrolled Solution [2+2] Polymerization of <italic style="font-style: italic">p</italic>-Phenylenediacrylate and Depolymerization](https://founder-rc-product.oss-cn-zhangjiakou.aliyuncs.com/upload/2025-10-30/102AWQ3DCRAEM0329690.png?response-content-type=image%2Fjpeg)
RESEARCH ARTICLE
2025, 43(11): 1981-1990. DOI: 10.1007/s10118-025-3418-3Published(online): 2025-10-30Abstract Full text L-PDFAbstract
RESEARCH ARTICLE
2025, 43(11): 1991-1999. DOI: 10.1007/s10118-025-3423-6Published(online): 2025-10-30Abstract Full text L-PDFAbstract
RESEARCH ARTICLE
2025, 43(11): 2000-2008. DOI: 10.1007/s10118-025-3441-4Published(online): 2025-10-30Abstract Full text L-PDFAbstract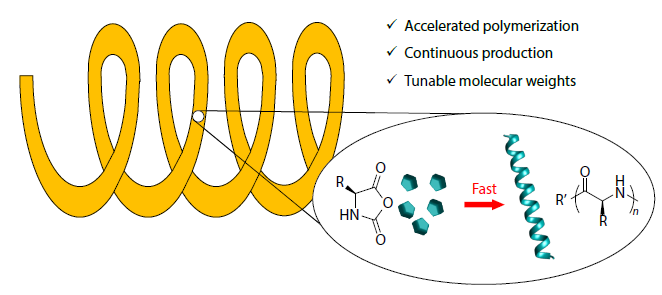
RESEARCH ARTICLE
2025, 43(11): 2009-2021. DOI: 10.1007/s10118-025-3427-2Published(online): 2025-10-30Abstract Full text L-PDFAbstract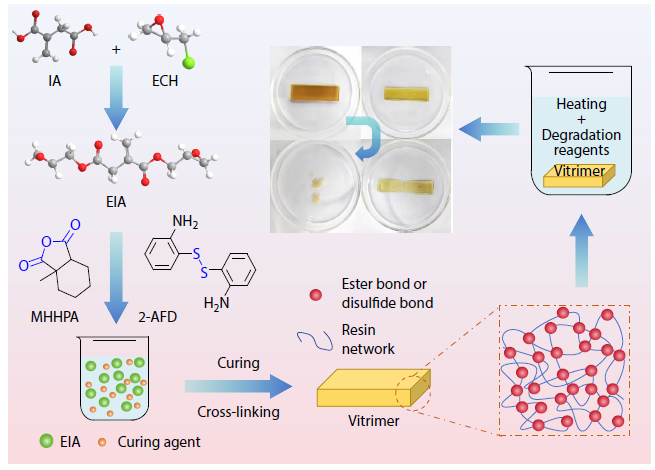
RESEARCH ARTICLE
2025, 43(11): 2022-2029. DOI: 10.1007/s10118-025-3414-7Published(online): 2025-10-30Abstract Full text L-PDFAbstract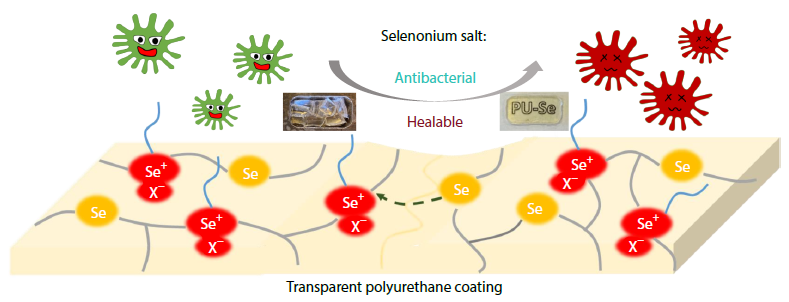
RESEARCH ARTICLE
2025, 43(11): 2030-2041. DOI: 10.1007/s10118-025-3413-8Published(online): 2025-10-30Abstract Full text L-PDFAbstract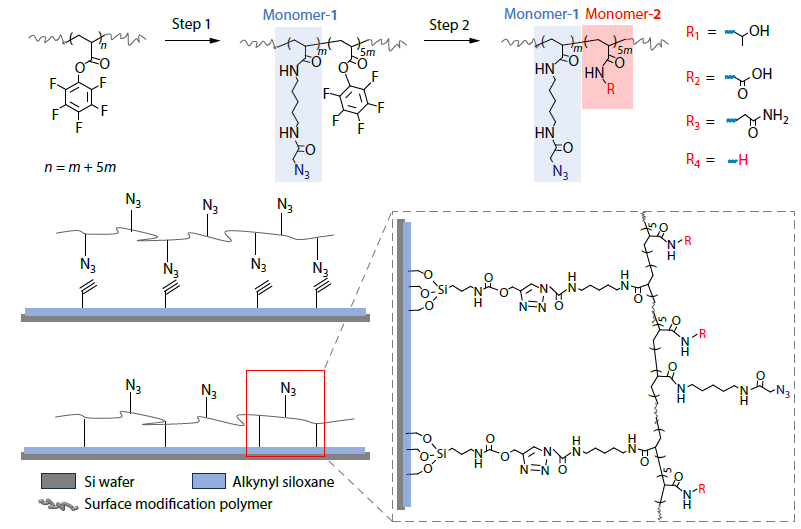
RESEARCH ARTICLE
2025, 43(11): 2042-2050. DOI: 10.1007/s10118-025-3411-xPublished(online): 2025-10-30Abstract Full text L-PDFAbstract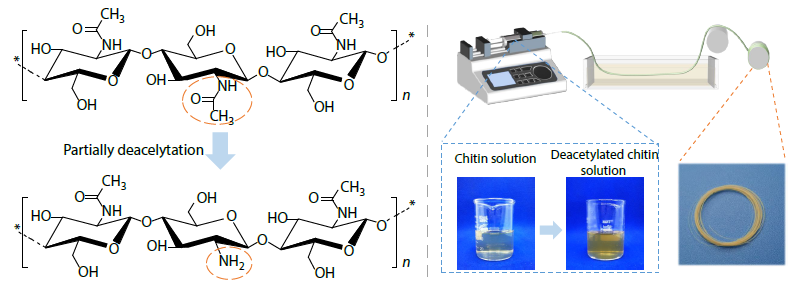
RESEARCH ARTICLE
2025, 43(11): 2051-2060. DOI: 10.1007/s10118-025-3429-0Published(online): 2025-10-30Abstract Full text L-PDFAbstract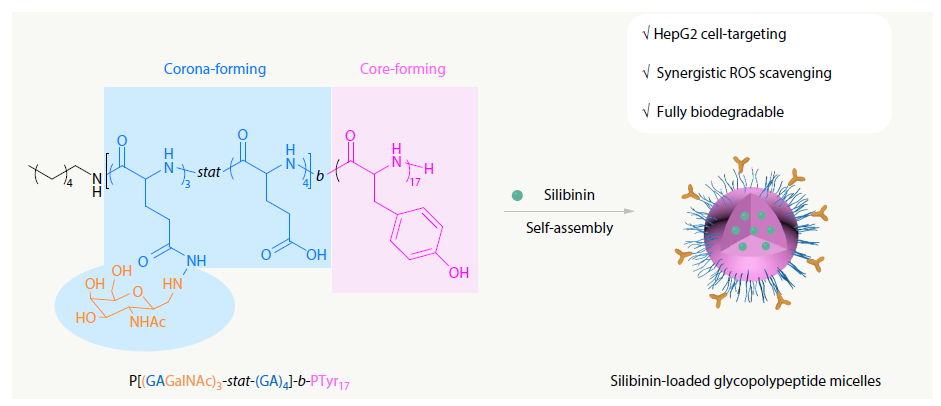
RESEARCH ARTICLE
2025, 43(11): 2061-2072. DOI: 10.1007/s10118-025-3422-7Published(online): 2025-10-30Abstract Full text L-PDFAbstract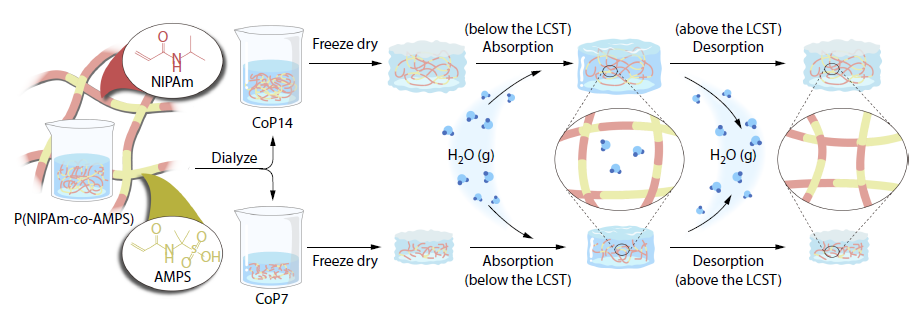
RESEARCH ARTICLE
2025, 43(11): 2073-2082. DOI: 10.1007/s10118-025-3417-4Published(online): 2025-10-30Abstract Full text L-PDFAbstract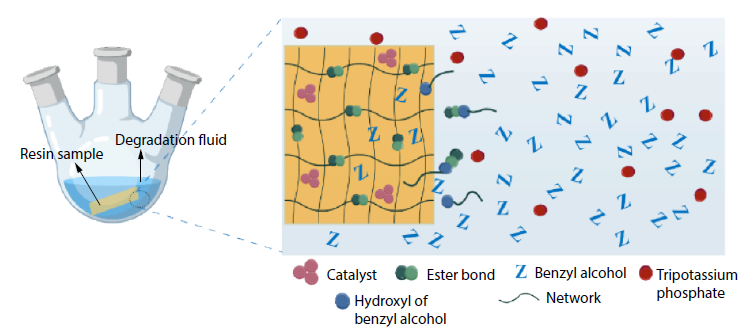
RESEARCH ARTICLE
2025, 43(11): 2083-2093. DOI: 10.1007/s10118-025-3419-2Published(online): 2025-10-30Abstract Full text L-PDFAbstract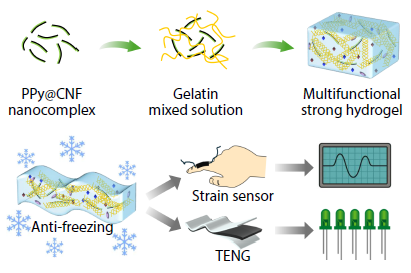
RESEARCH ARTICLE
2025, 43(11): 2094-2101. DOI: 10.1007/s10118-025-3420-9Published(online): 2025-10-30Abstract Full text L-PDFAbstract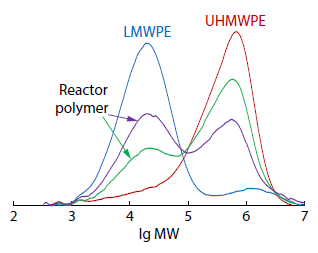
RESEARCH ARTICLE
2025, 43(11): 2102-2109. DOI: 10.1007/s10118-025-3433-4Published(online): 2025-10-30Abstract Full text L-PDFAbstract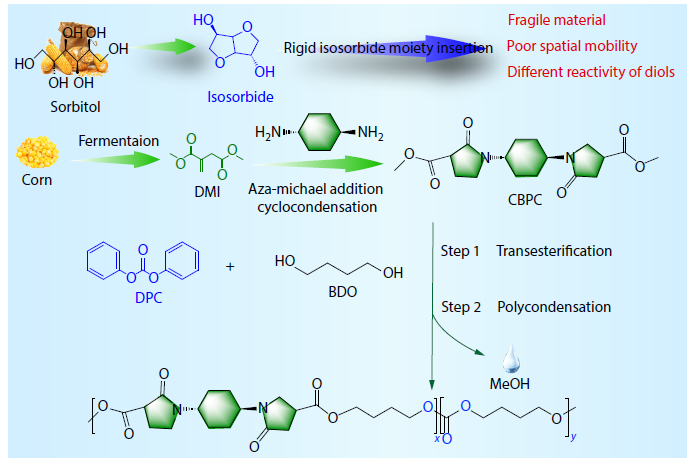
RESEARCH ARTICLE
2025, 43(11): 2110-2117. DOI: 10.1007/s10118-025-3425-4Published(online): 2025-10-30Abstract Full text L-PDFAbstract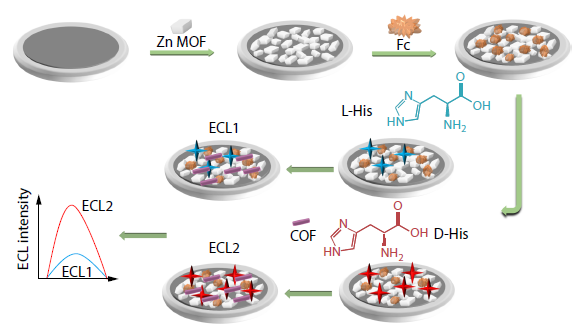
RESEARCH ARTICLE
2025, 43(11): 2118-2127. DOI: 10.1007/s10118-025-3428-1Published(online): 2025-10-30Abstract Full text L-PDFAbstract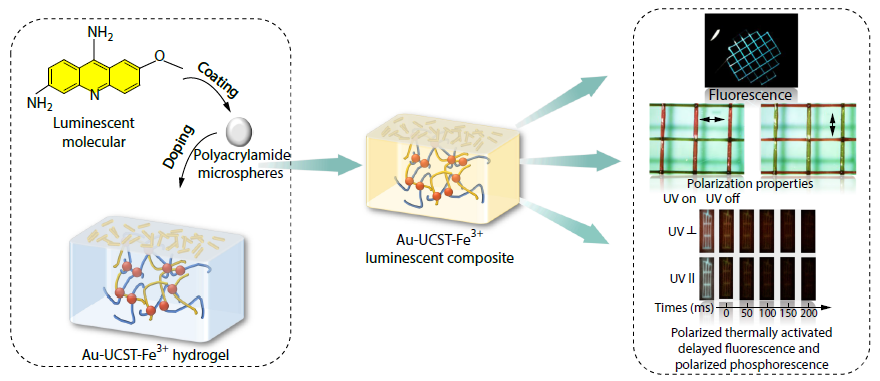
RESEARCH ARTICLE
2025, 43(11): 2128-2137. DOI: 10.1007/s10118-025-3412-9Published(online): 2025-10-30Abstract Full text L-PDFAbstract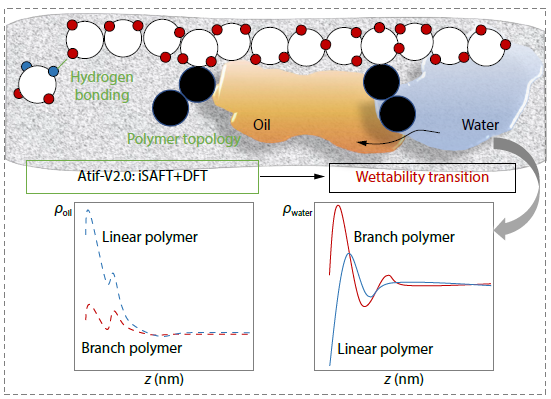
RESEARCH ARTICLE
2025, 43(11): 2138-2149. DOI: 10.1007/s10118-025-3405-8Published(online): 2025-10-30Abstract Full text L-PDFAbstract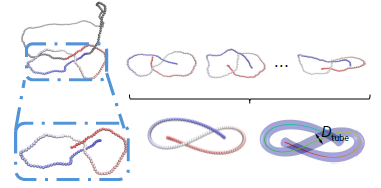
RESEARCH ARTICLE
2025, 43(11): 2150-2159. DOI: 10.1007/s10118-025-3421-8Published(online): 2025-10-30Abstract Full text L-PDFAbstract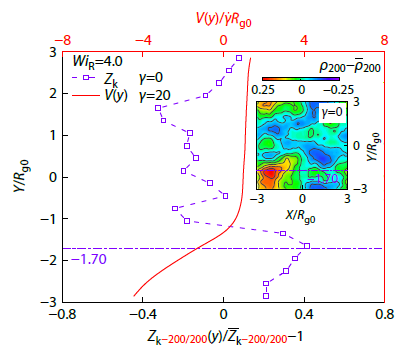
RESEARCH ARTICLE
2025, 43(11): 2160-2170. DOI: 10.1007/s10118-025-3375-xPublished(online): 2025-10-30Abstract Full text L-PDFAbstract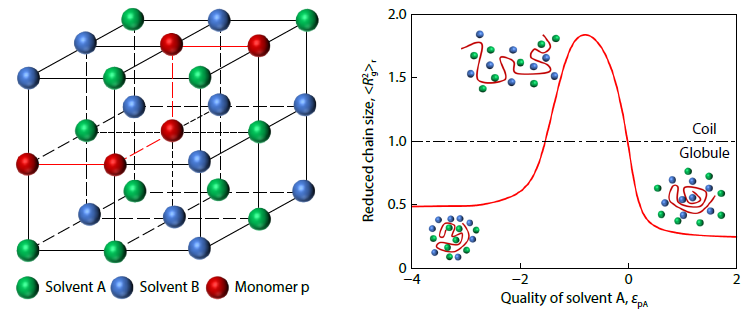
RESEARCH ARTICLE
2025, 43(11): 2171-2184. DOI: 10.1007/s10118-025-3416-5Published(online): 2025-10-30Abstract Full text L-PDFAbstract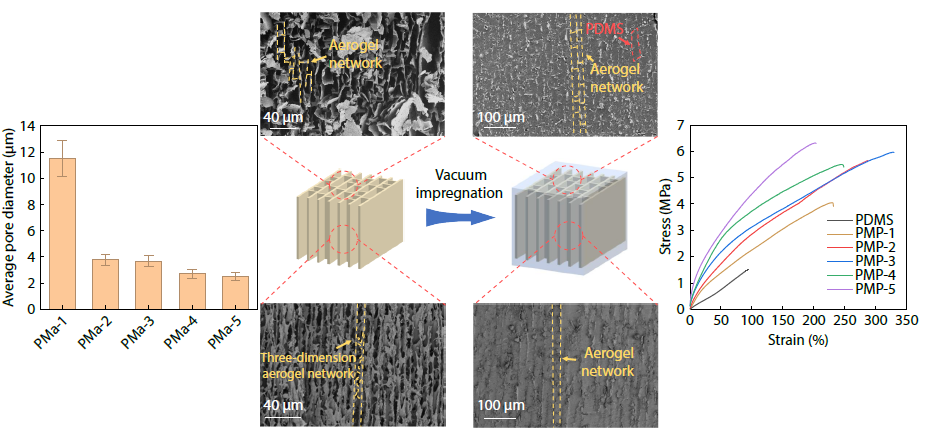
- Technical support is provided by Beijing Founder electronics co., LTD 京ICP备09064830号-19
 京公网安备11010802024621
京公网安备11010802024621 - It is recommended to read the content of this site in Chrome&IE9+. Please switch to extreme mode in browser 360.
- Cookies We use cookies to help provide and enhance our service and tailor content. By continuing, you agree to the use of cookies.
0