
Latest Issue
Just Accepted Online First List of Issues Document Types
Issue 12, 2021
REVIEW
2021, 39(12): 1521-1527. DOI: 10.1007/s10118-021-2615-yPublished(online): 2021-07-25Abstract Full text L-PDFAbstract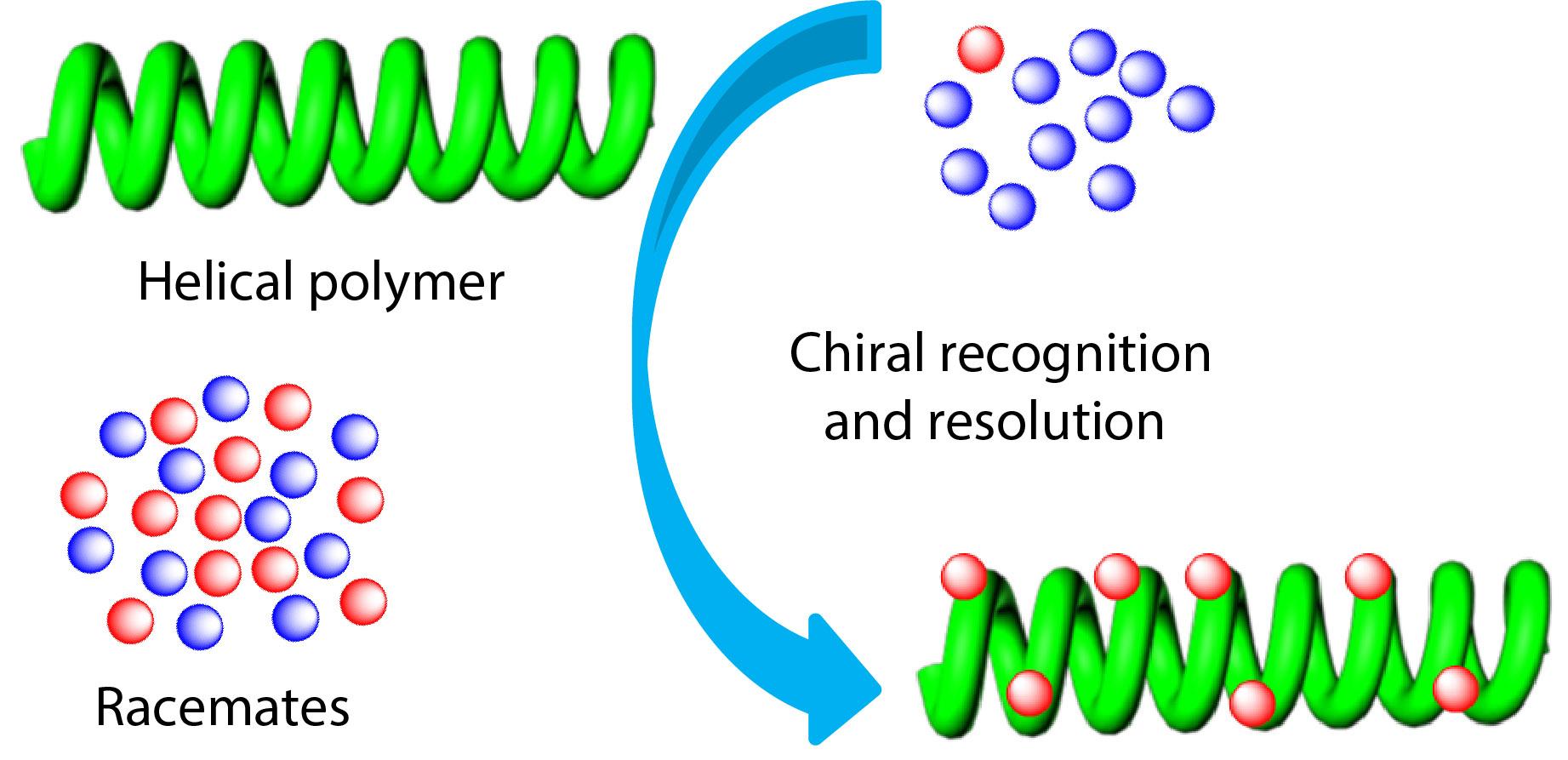

ARTICLE
2021, 39(12): 1528-1537. DOI: 10.1007/s10118-021-2580-5Published(online): 2021-06-04Abstract Full text L-PDFAbstract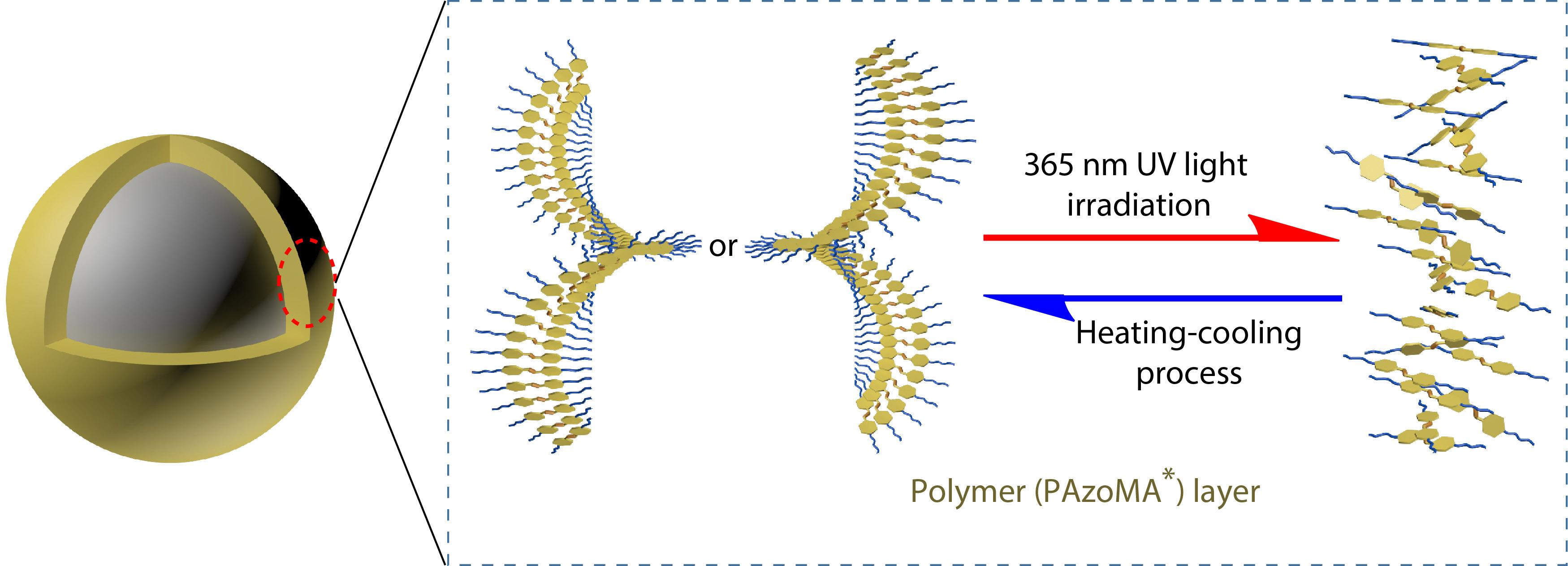
ARTICLE
2021, 39(12): 1538-1549. DOI: 10.1007/s10118-021-2599-7Published(online): 2021-06-18Abstract Full text L-PDFAbstract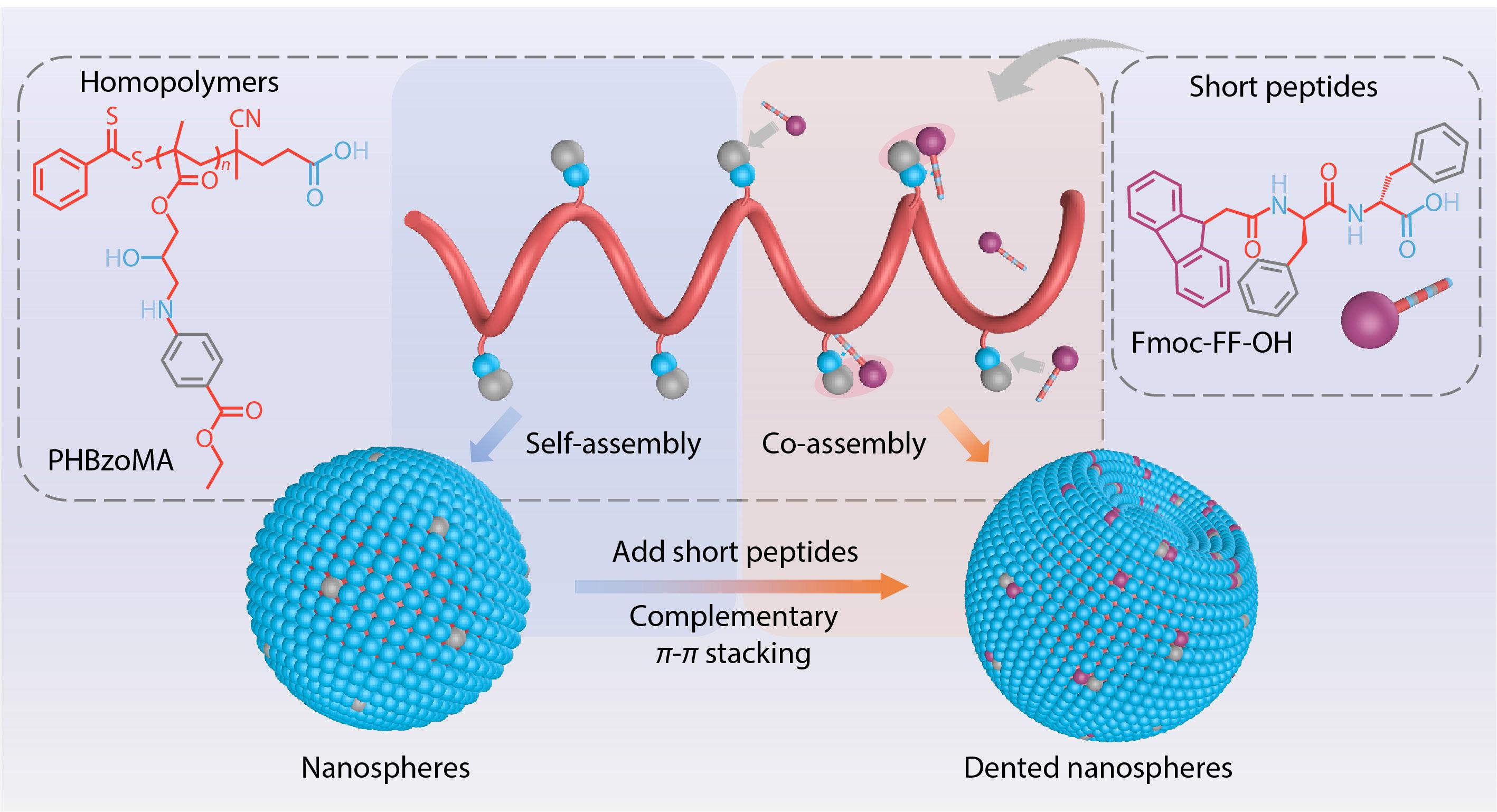
ARTICLE
2021, 39(12): 1550-1561. DOI: 10.1007/s10118-021-2621-0Published(online): 2021-08-26Abstract Full text L-PDFAbstract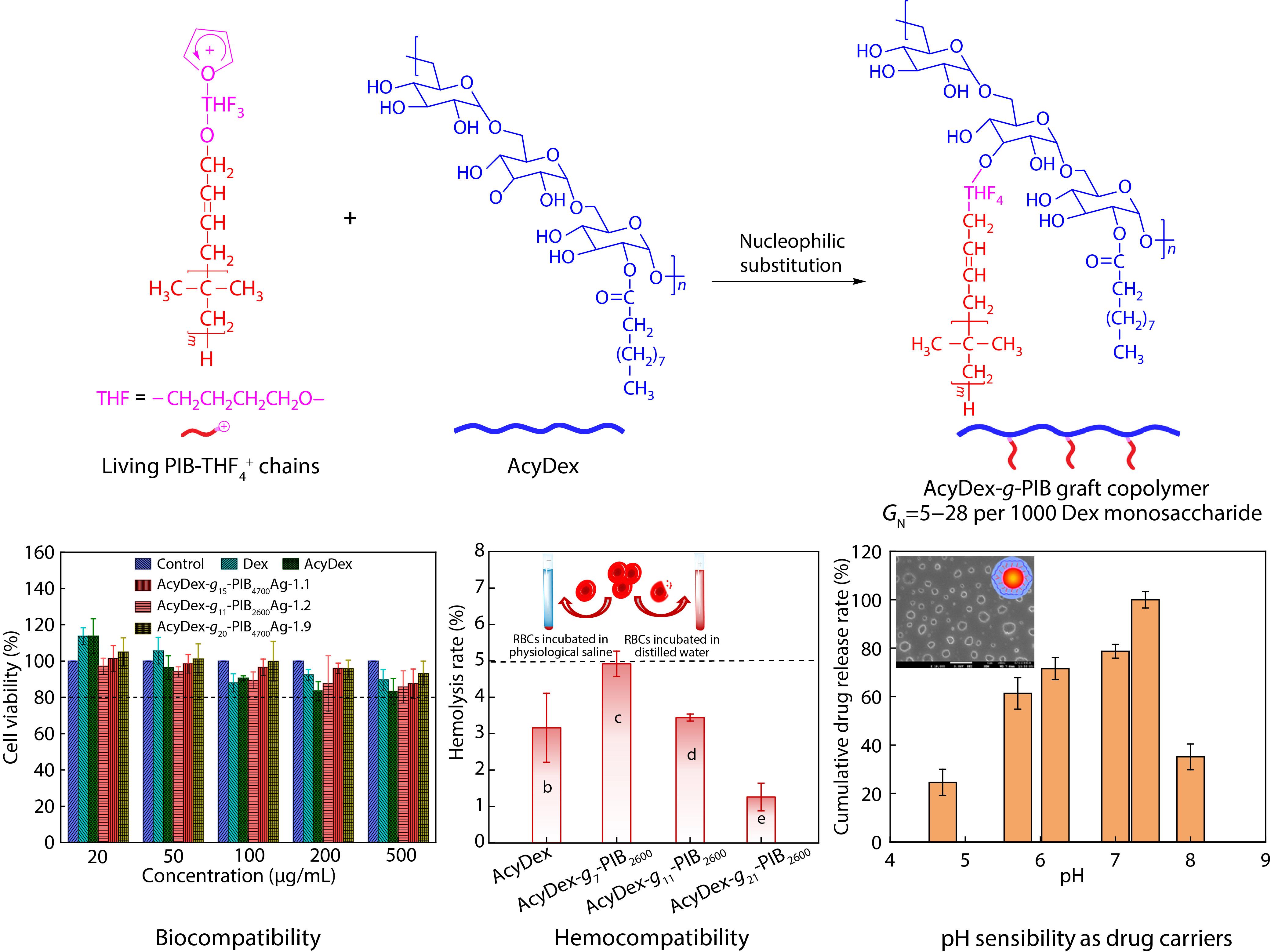
ARTICLE
2021, 39(12): 1562-1571. DOI: 10.1007/s10118-021-2598-8Published(online): 2021-07-06Abstract Full text L-PDFAbstract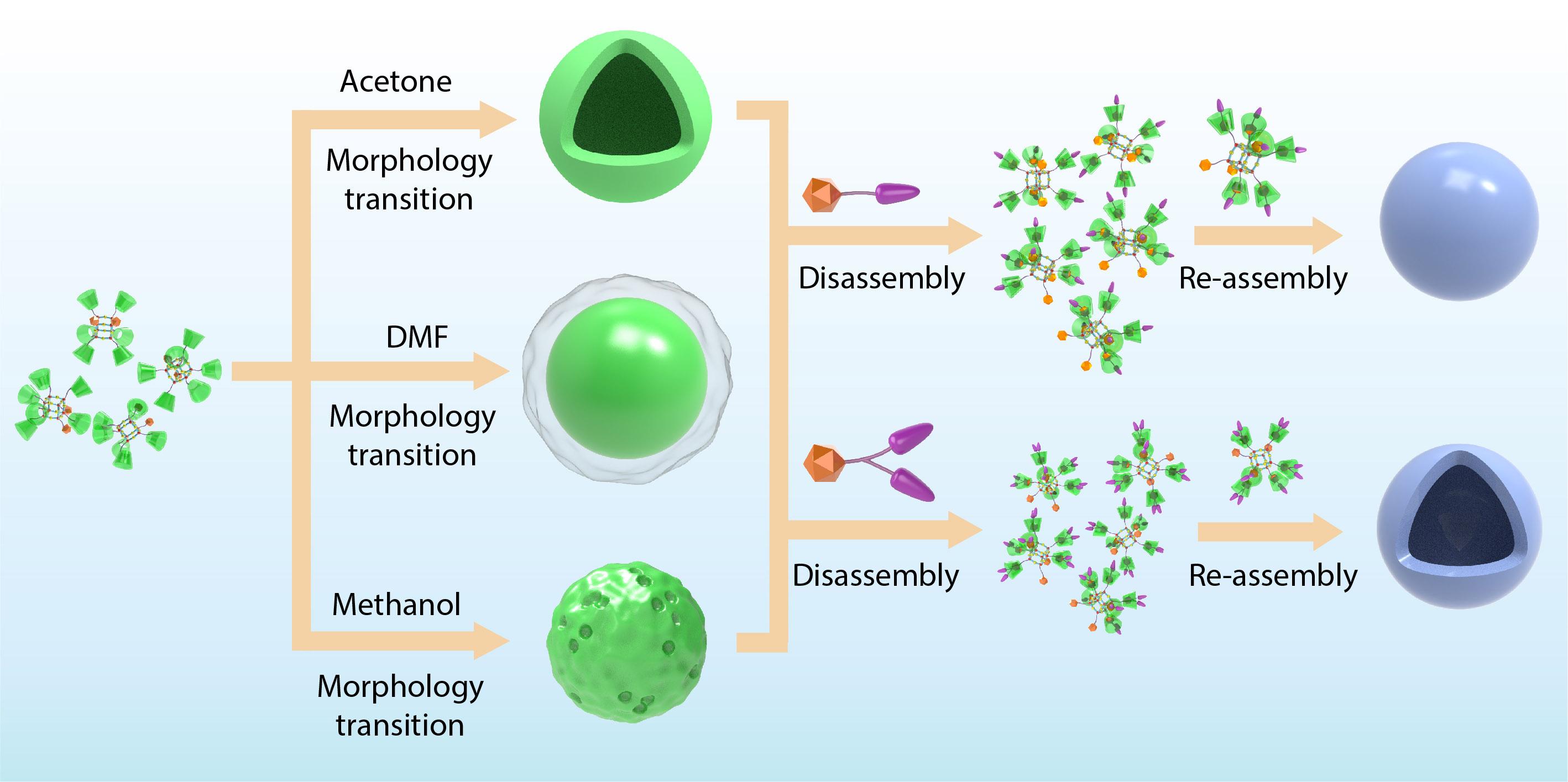
ARTICLE
2021, 39(12): 1572-1580. DOI: 10.1007/s10118-021-2590-3Published(online): 2021-06-16Abstract Full text L-PDFAbstract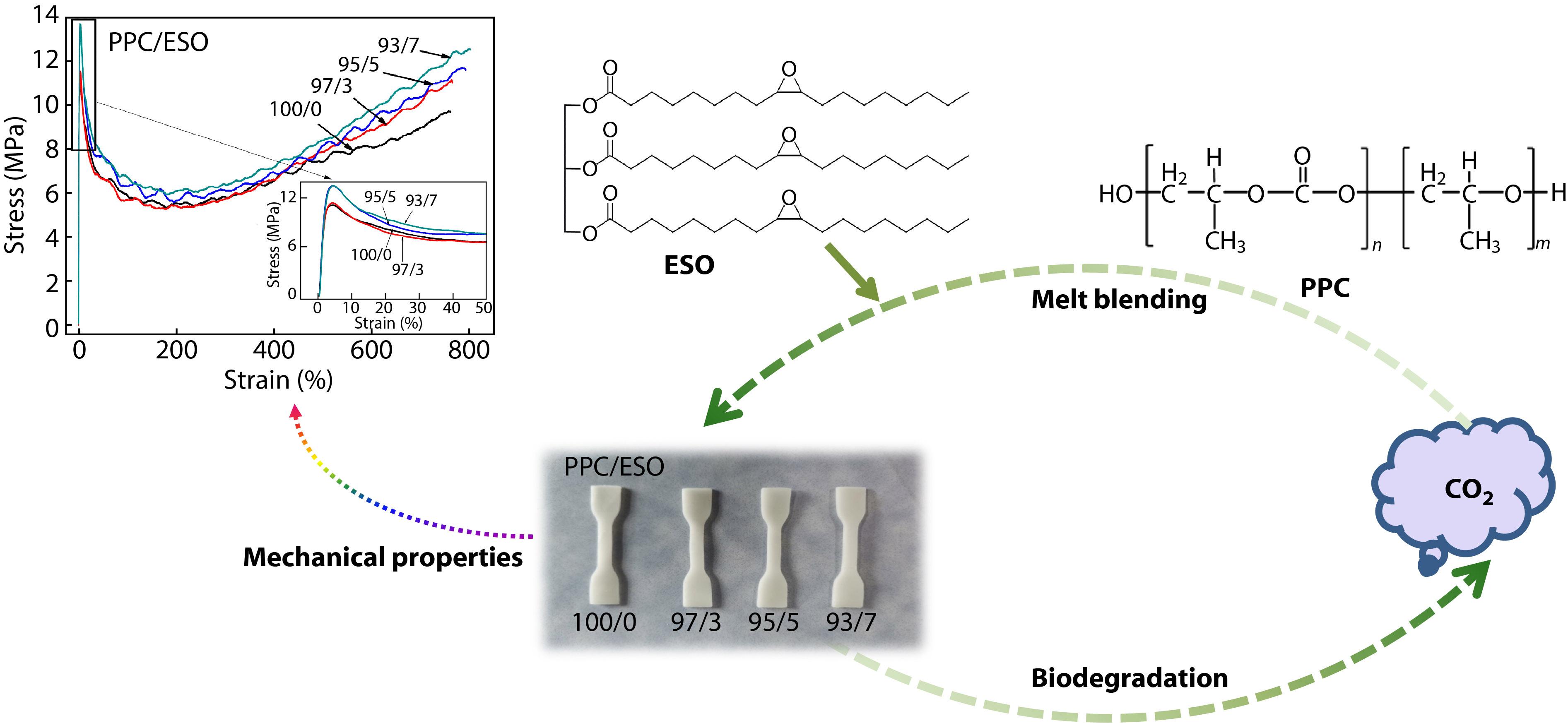
ARTICLE
2021, 39(12): 1581-1589. DOI: 10.1007/s10118-021-2629-5Published(online): 2021-08-26Abstract Full text L-PDFAbstract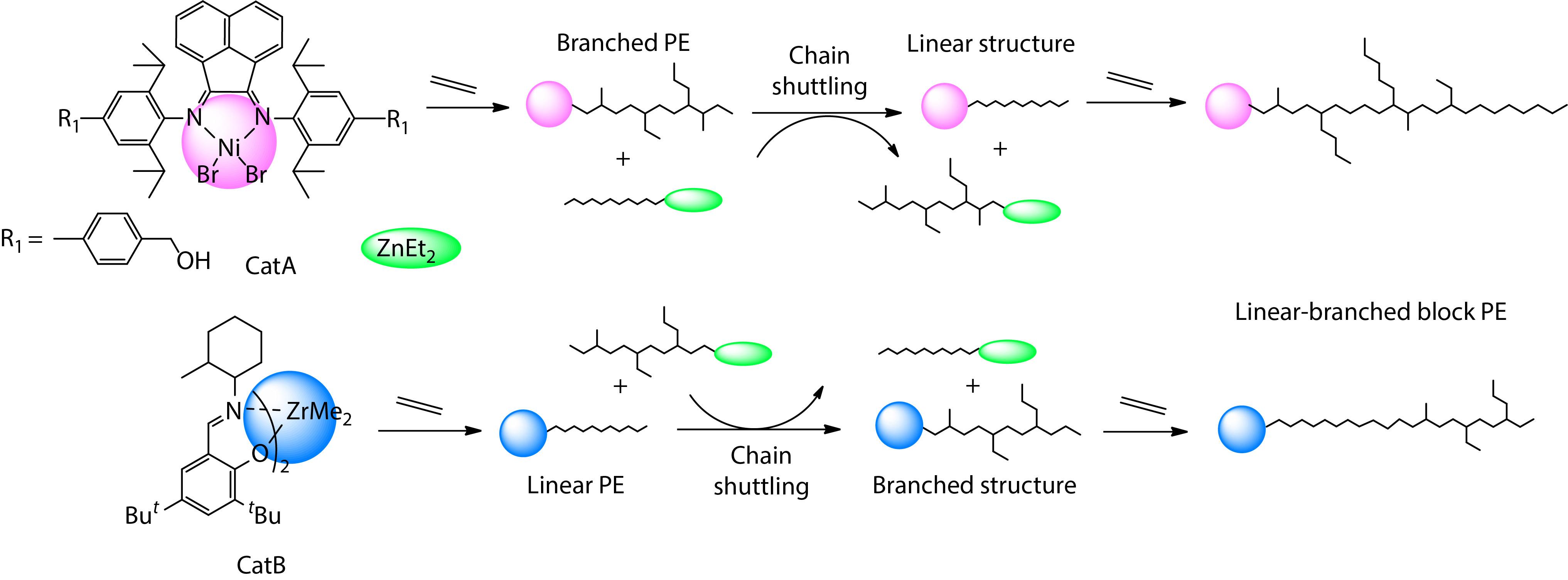
ARTICLE
2021, 39(12): 1590-1596. DOI: 10.1007/s10118-021-2605-0Published(online): 2021-07-23Abstract Full text L-PDFAbstract
ARTICLE
2021, 39(12): 1597-1608. DOI: 10.1007/s10118-021-2613-0Published(online): 2021-07-30Abstract Full text L-PDFAbstract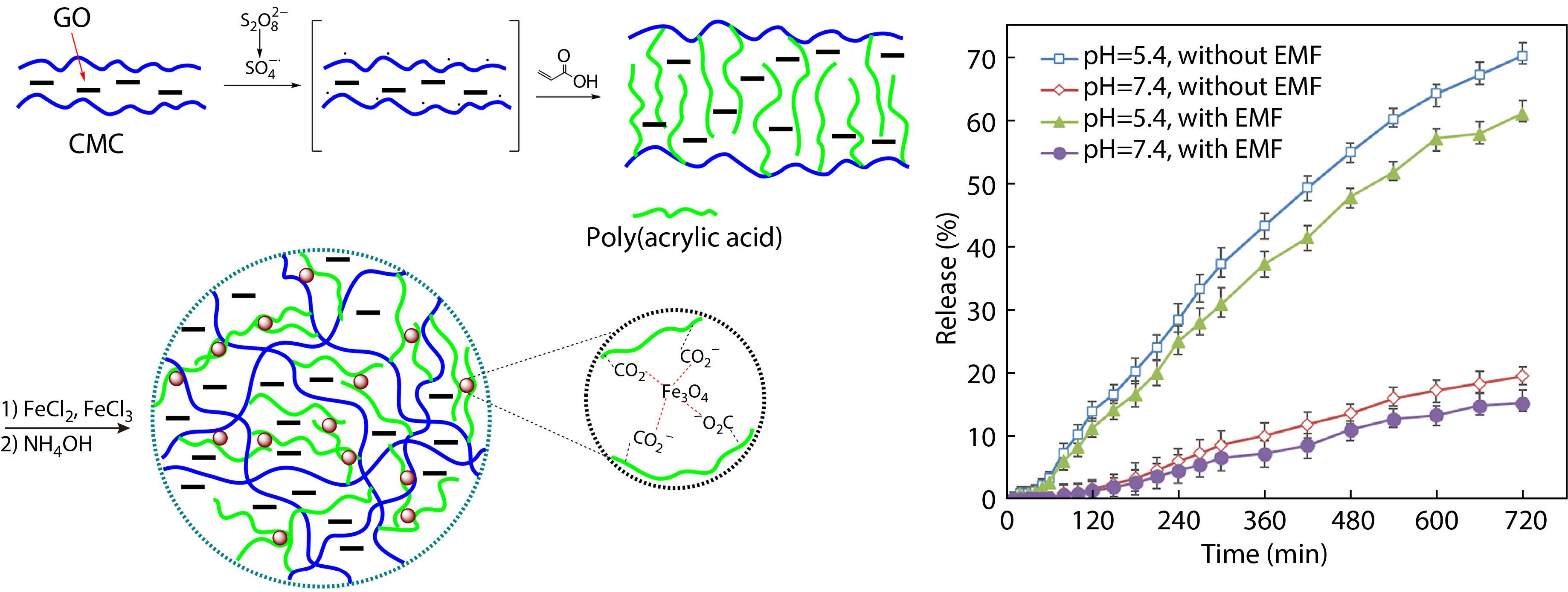
ARTICLE
2021, 39(12): 1609-1616. DOI: 10.1007/s10118-021-2617-9Published(online): 2021-07-30Abstract Full text L-PDFAbstract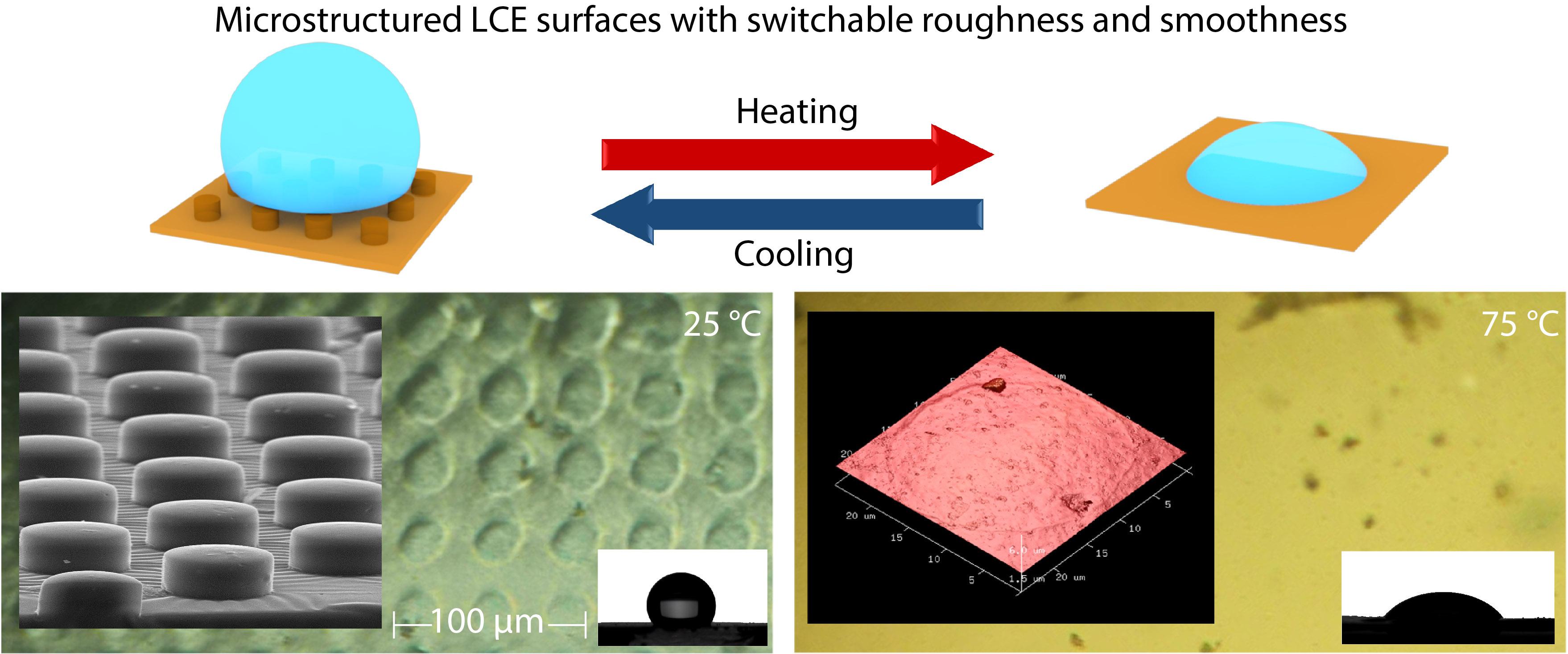
ARTICLE
2021, 39(12): 1617-1625. DOI: 10.1007/s10118-021-2618-8Published(online): 2021-07-30Abstract Full text L-PDFAbstract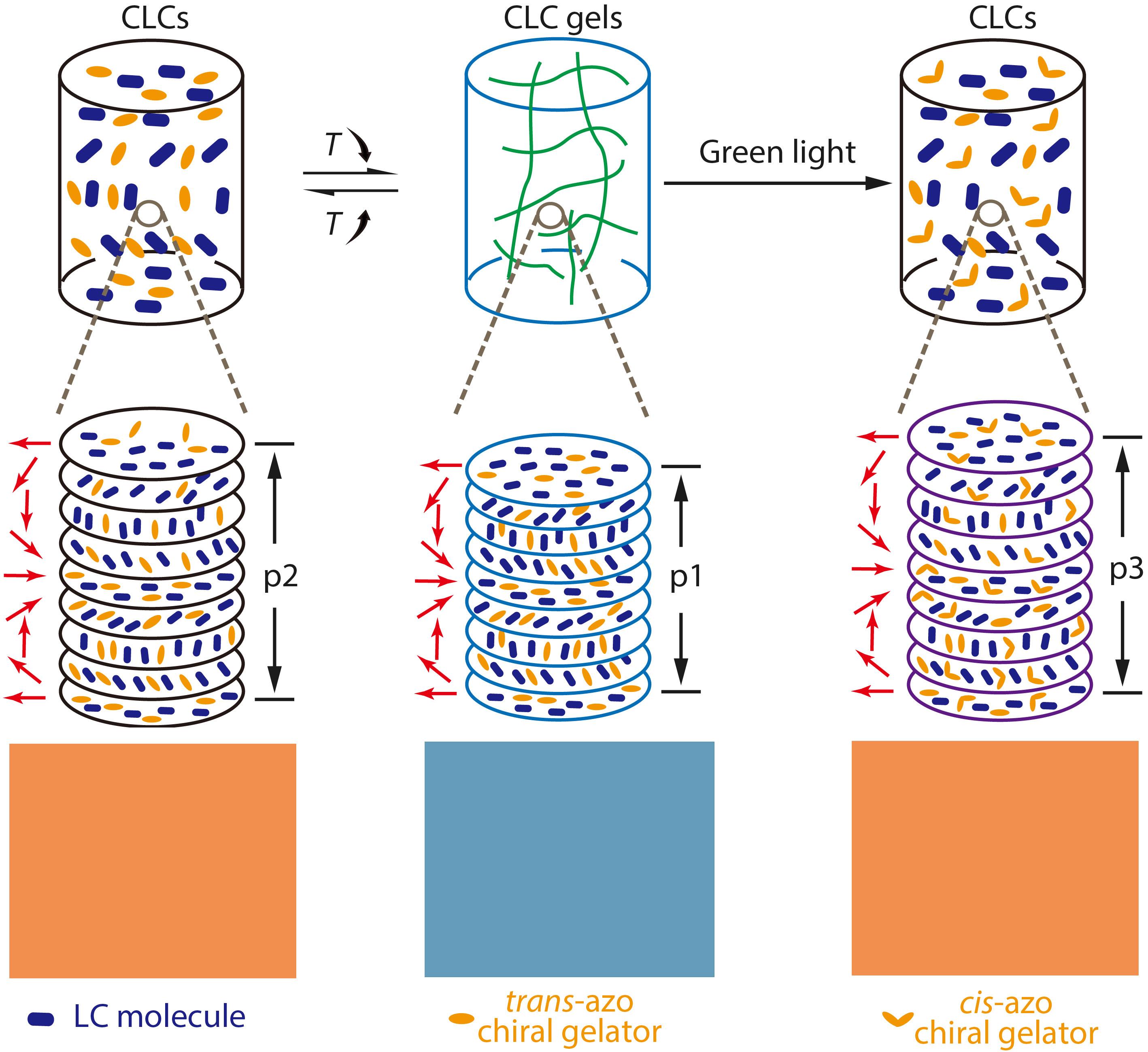
ARTICLE
2021, 39(12): 1626-1633. DOI: 10.1007/s10118-021-2614-zPublished(online): 2021-07-23Abstract Full text L-PDFAbstract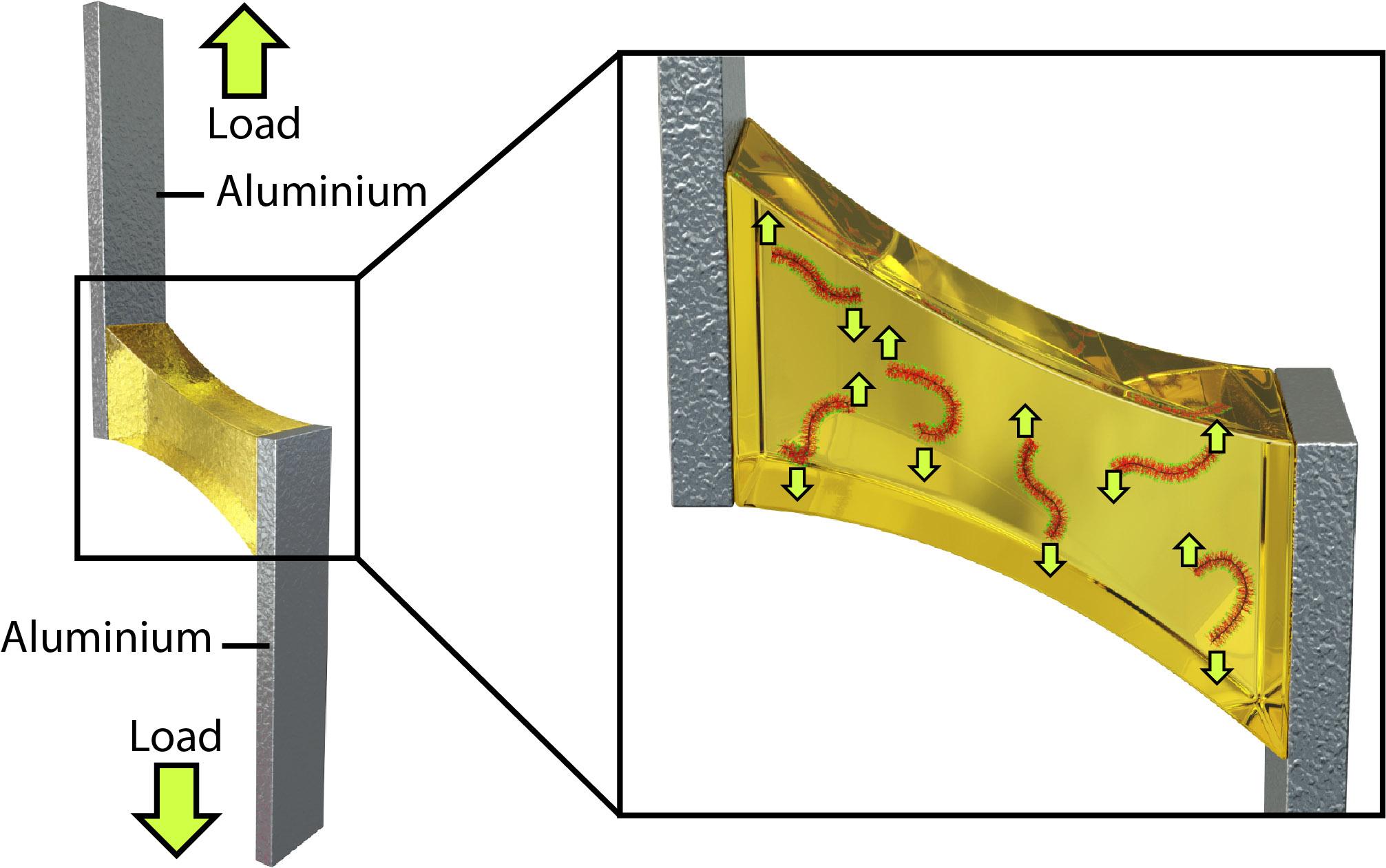
ARTICLE
2021, 39(12): 1634-1644. DOI: 10.1007/s10118-021-2634-8Published(online): 2021-09-06Abstract Full text L-PDFAbstract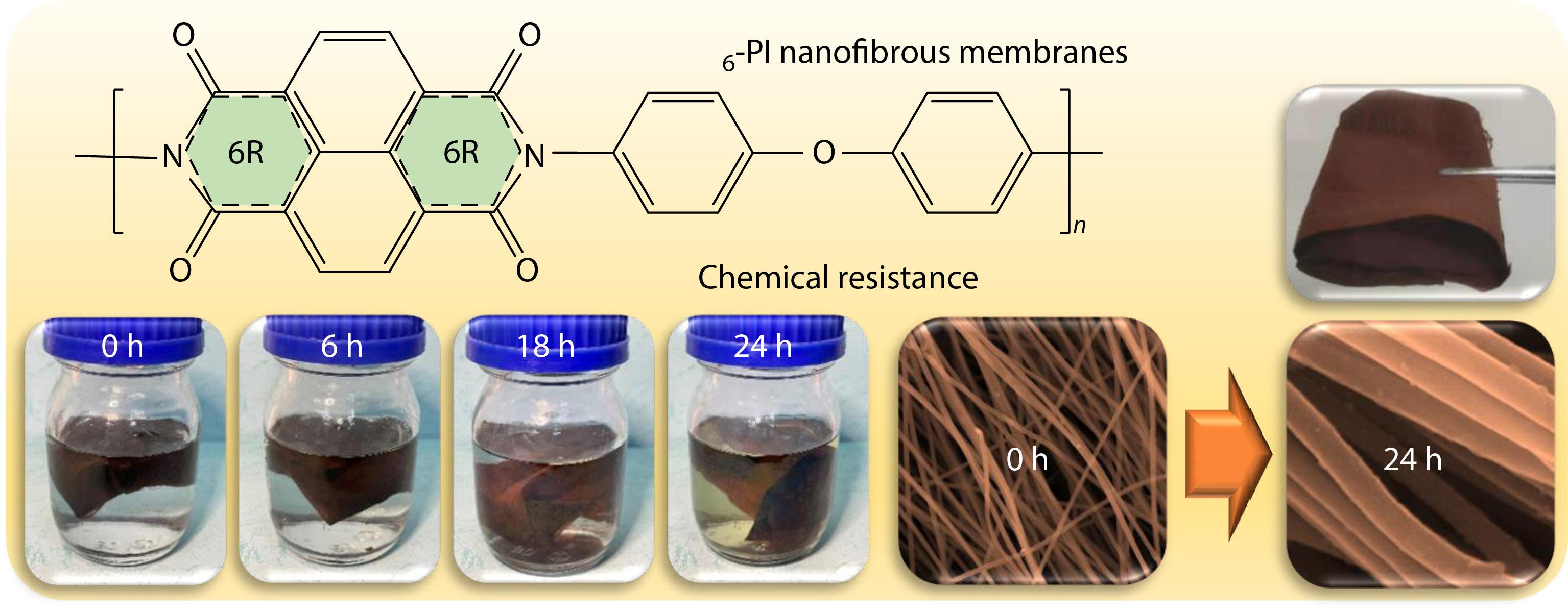
ARTICLE
2021, 39(12): 1645-1656. DOI: 10.1007/s10118-021-2632-xPublished(online): 2021-09-06Abstract Full text L-PDFAbstract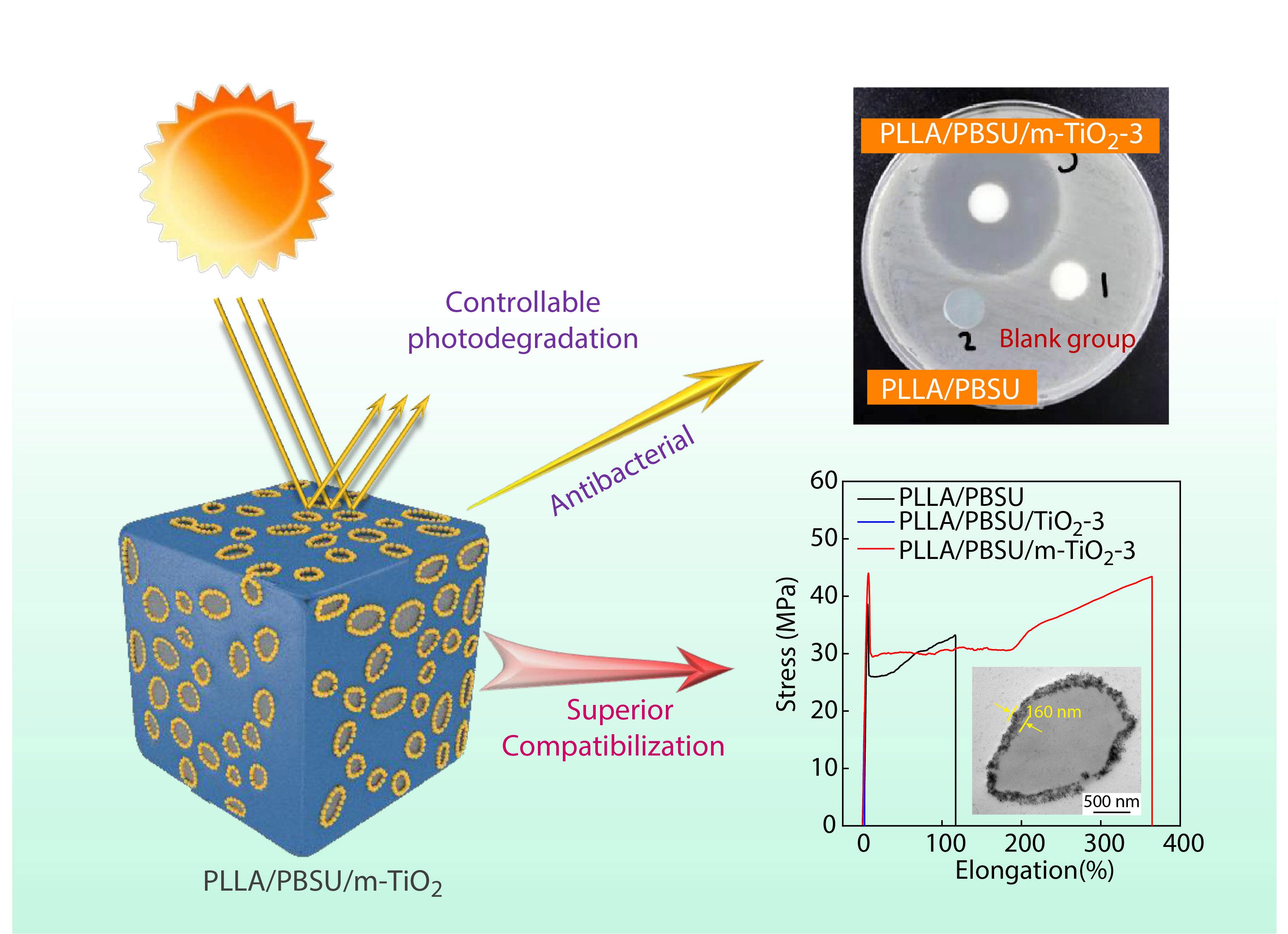
ARTICLE
2021, 39(12): 1657-1664. DOI: 10.1007/s10118-021-2619-7Published(online): 2021-07-30Abstract Full text L-PDFAbstract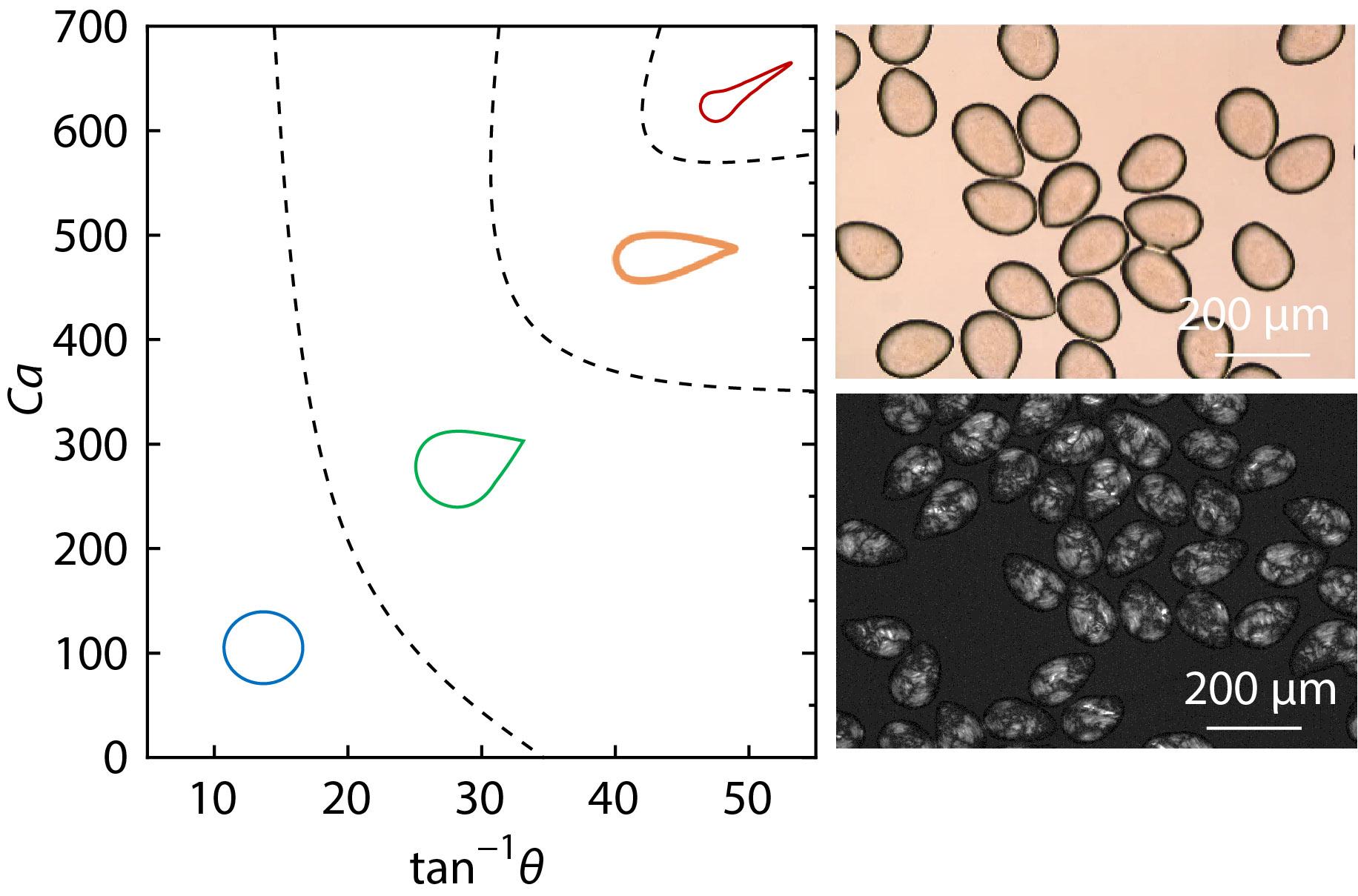
ARTICLE
2021, 39(12): 1665-1672. DOI: 10.1007/s10118-021-2628-6Published(online): 2021-08-20Abstract Full text L-PDFAbstract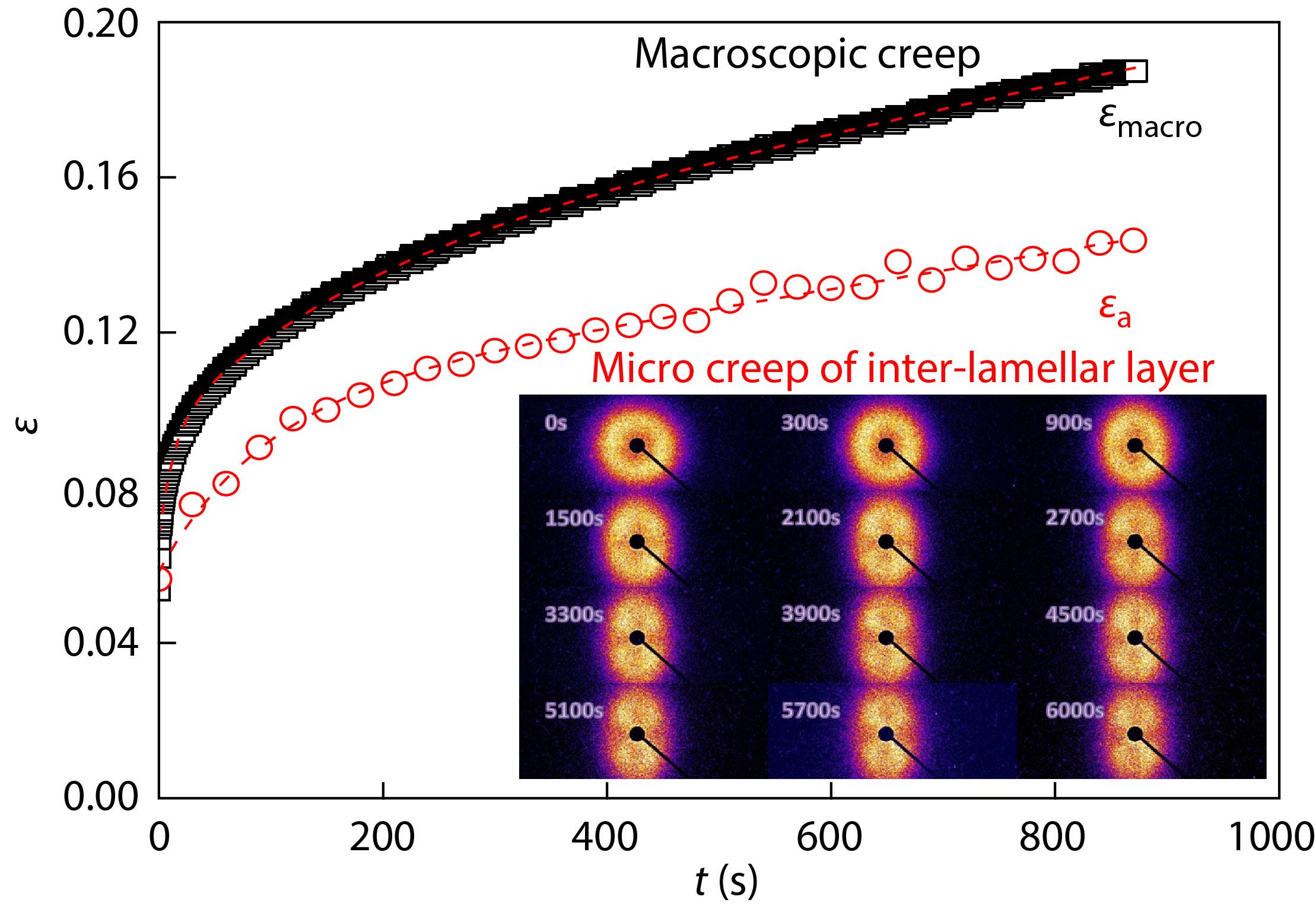
ARTICLE
2021, 39(12): 1673-1679. DOI: 10.1007/s10118-021-2600-5Published(online): 2021-07-05Abstract Full text L-PDFAbstract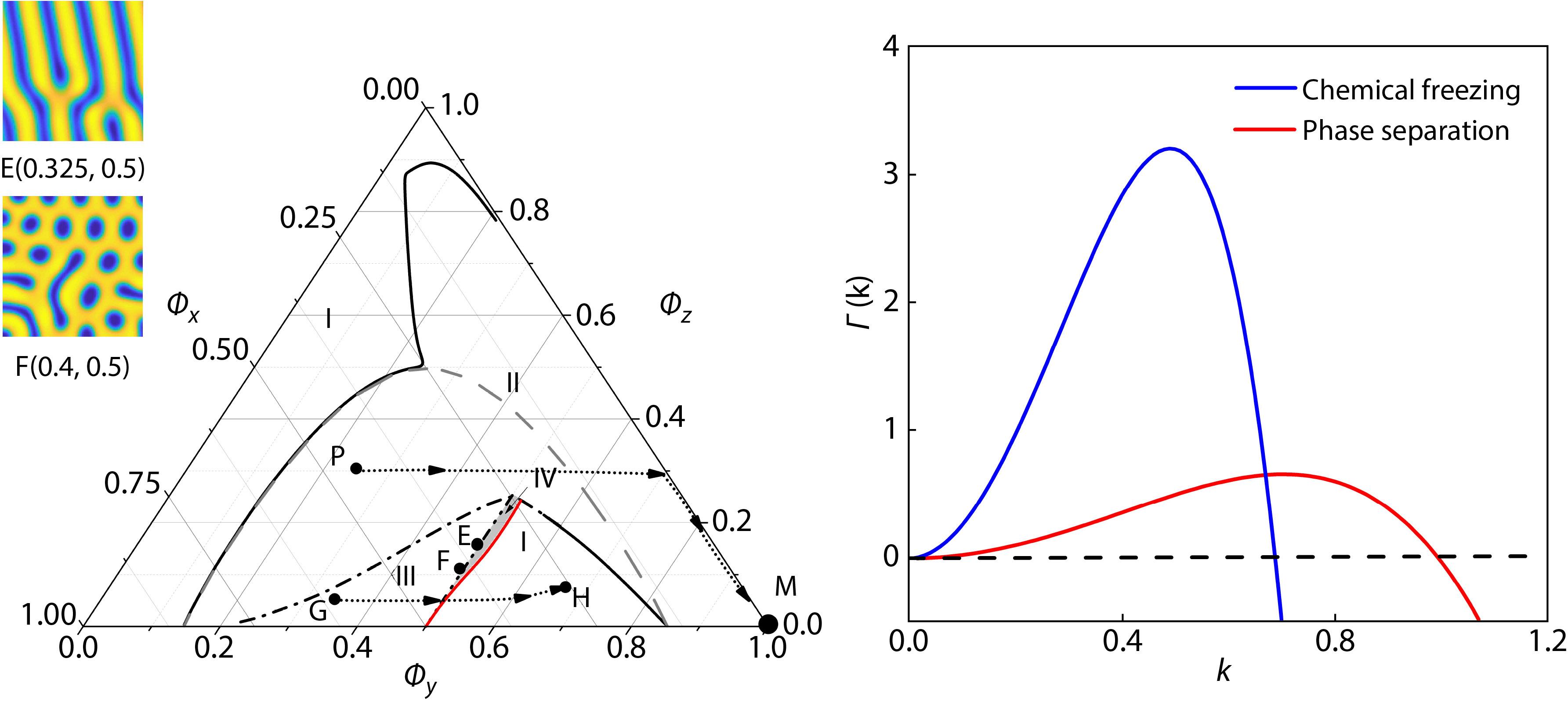
ARTICLE
2021, 39(12): 1680-1694. DOI: 10.1007/s10118-021-2606-zPublished(online): 2021-07-23Abstract Full text L-PDFAbstract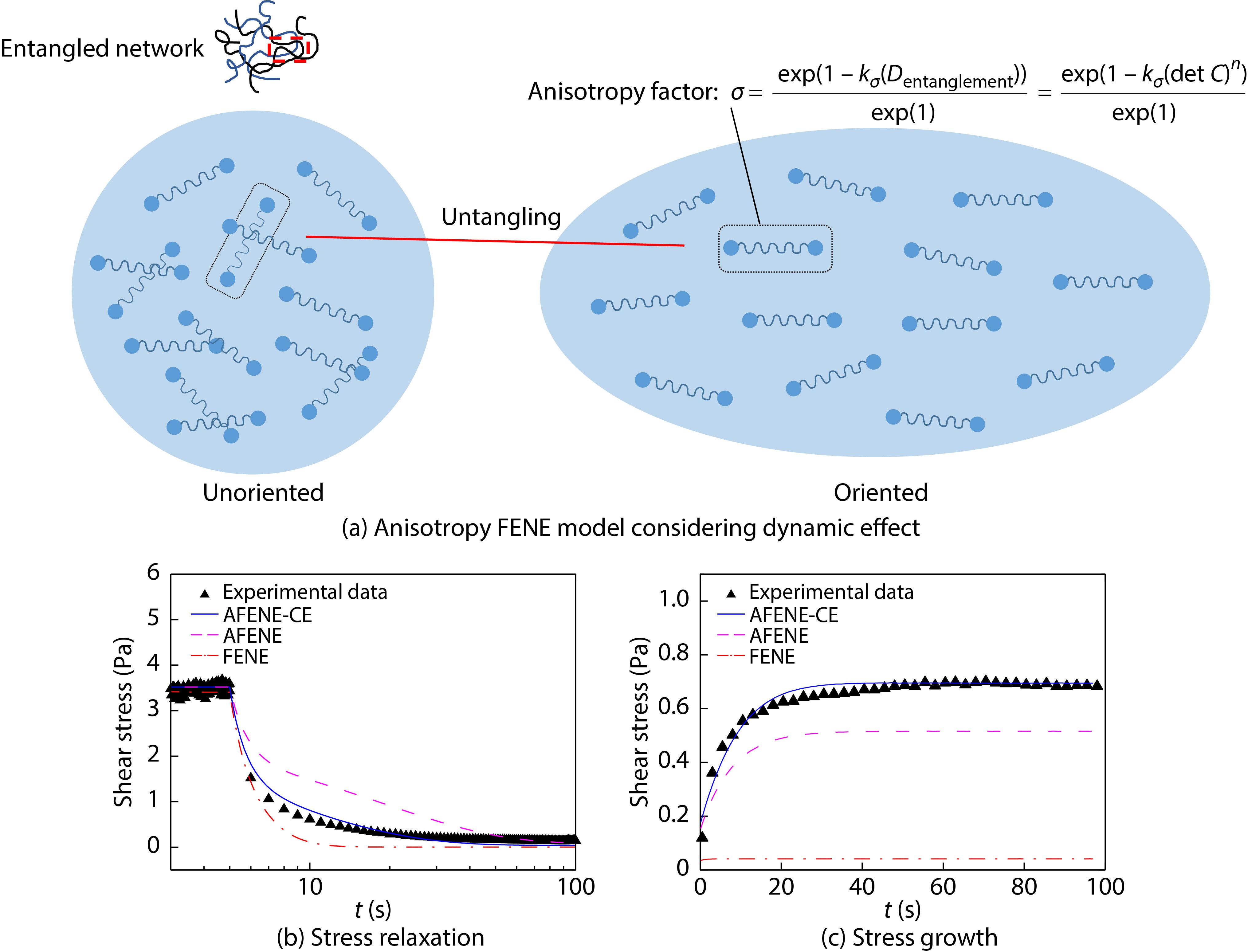
- Technical support is provided by Beijing Founder electronics co., LTD 京ICP备09064830号-19
 京公网安备11010802024621
京公网安备11010802024621 - It is recommended to read the content of this site in Chrome&IE9+. Please switch to extreme mode in browser 360.
- Cookies We use cookies to help provide and enhance our service and tailor content. By continuing, you agree to the use of cookies.
0