Issue Ebook
cover story

Latest Issue
Just Accepted Online First List of Issues Document Types
Issue 7, 2026
Special Topic: Distinguished Young Scholars in Polymer Science
2026, 44(7): 1957-1958. DOI: 10.1007/s10118-026-3786-3Published(online): 2026-07-01Full text L-PDFSpecial Topic: Distinguished Young Scholars in Polymer Science
2026, 44(7): 1959-1983. DOI: 10.1007/s10118-025-3545-xPublished(online): 2026-07-01Abstract Full text L-PDFAbstract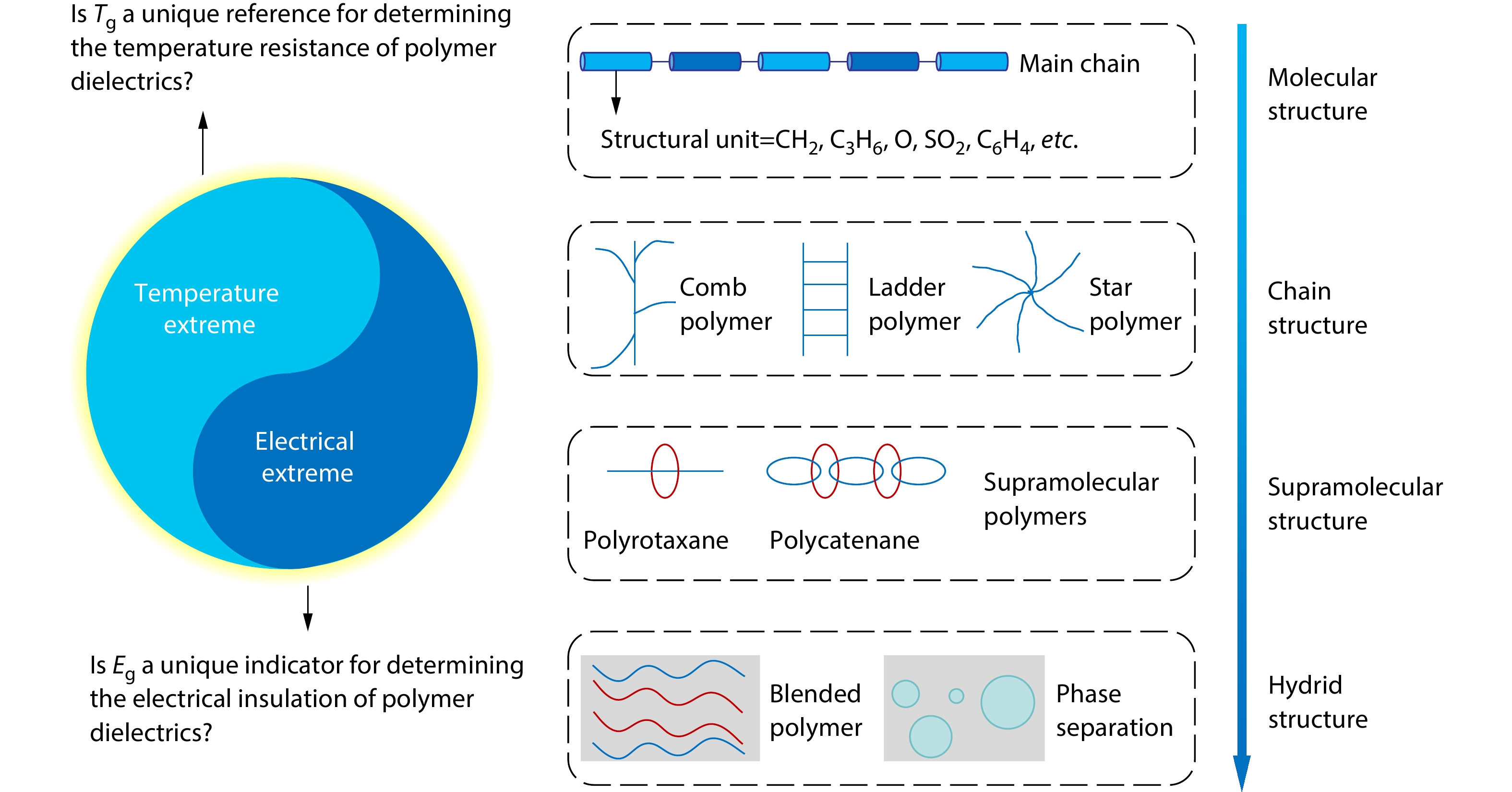
Special Topic: Distinguished Young Scholars in Polymer Science
2026, 44(7): 1984-2001. DOI: 10.1007/s10118-026-3567-zPublished(online): 2026-07-01Abstract Full text L-PDFAbstract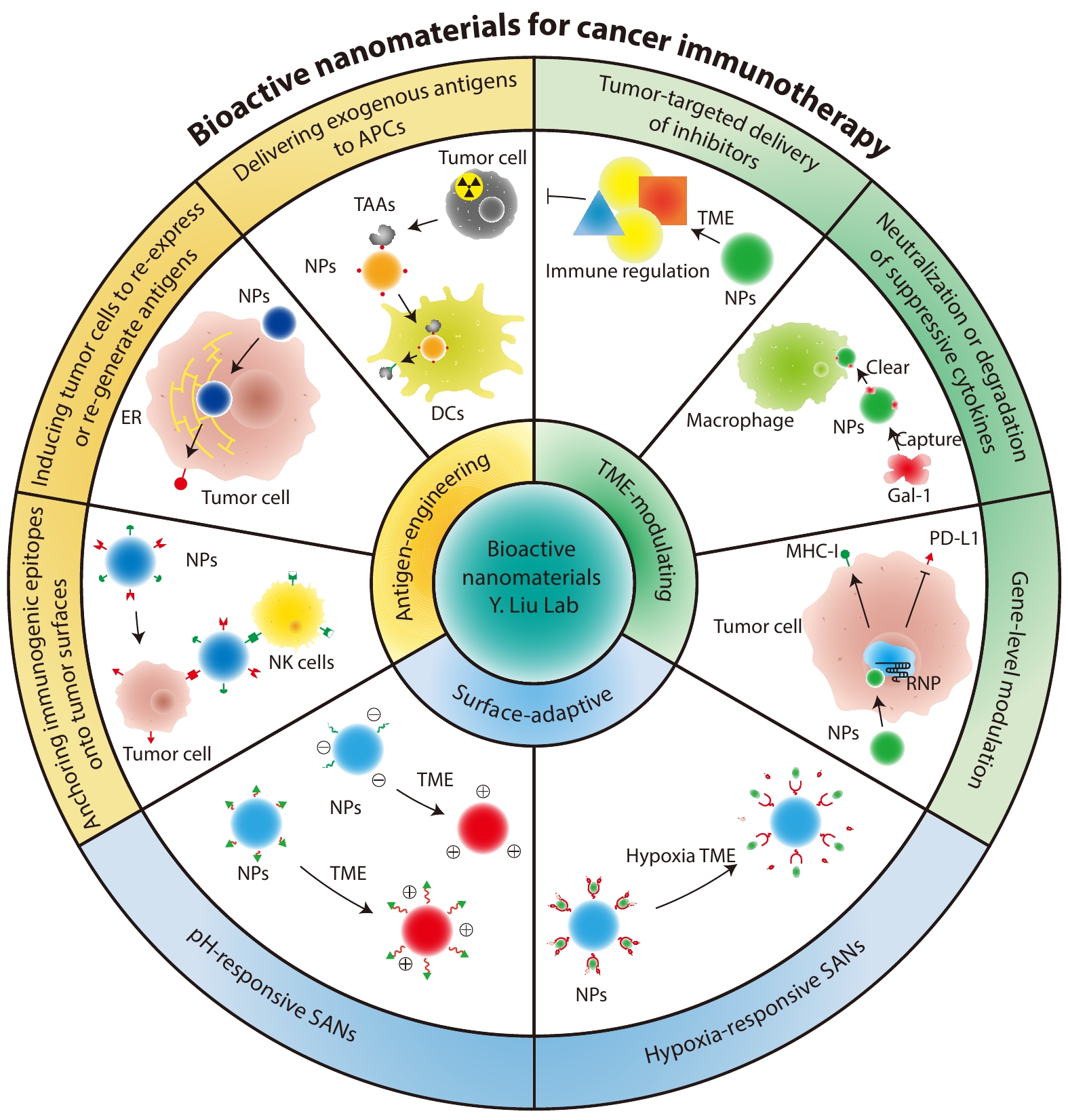
Special Topic: Distinguished Young Scholars in Polymer Science
2026, 44(7): 2002-2033. DOI: 10.1007/s10118-026-3652-3Published(online): 2026-07-01Abstract Full text L-PDFAbstract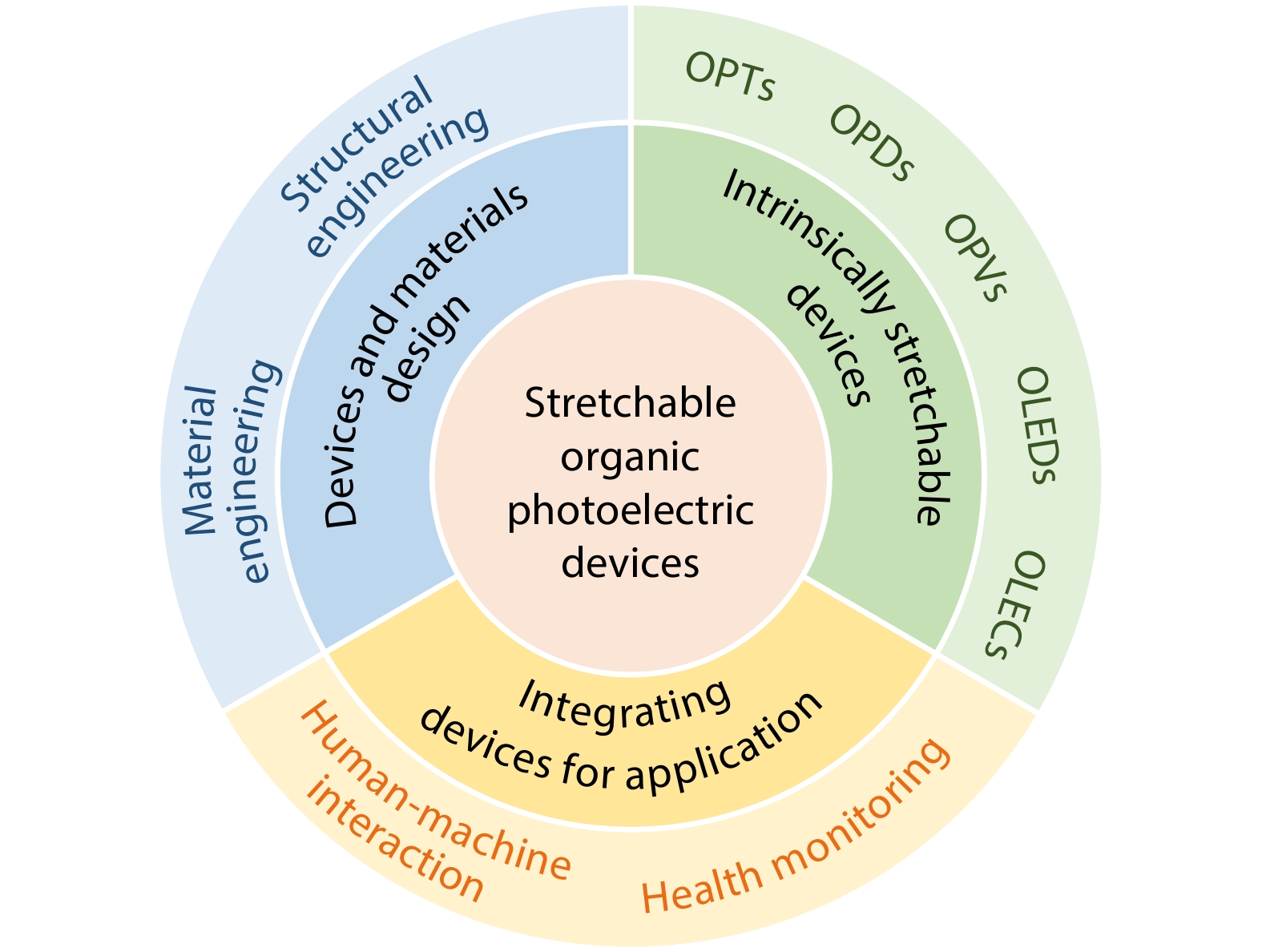
Special Topic: Distinguished Young Scholars in Polymer Science
2026, 44(7): 2034-2042. DOI: 10.1007/s10118-025-3499-zPublished(online): 2026-07-01Abstract Full text L-PDFAbstract
Special Topic: Distinguished Young Scholars in Polymer Science
2026, 44(7): 2043-2050. DOI: 10.1007/s10118-026-3666-xPublished(online): 2026-07-01Abstract Full text L-PDFAbstract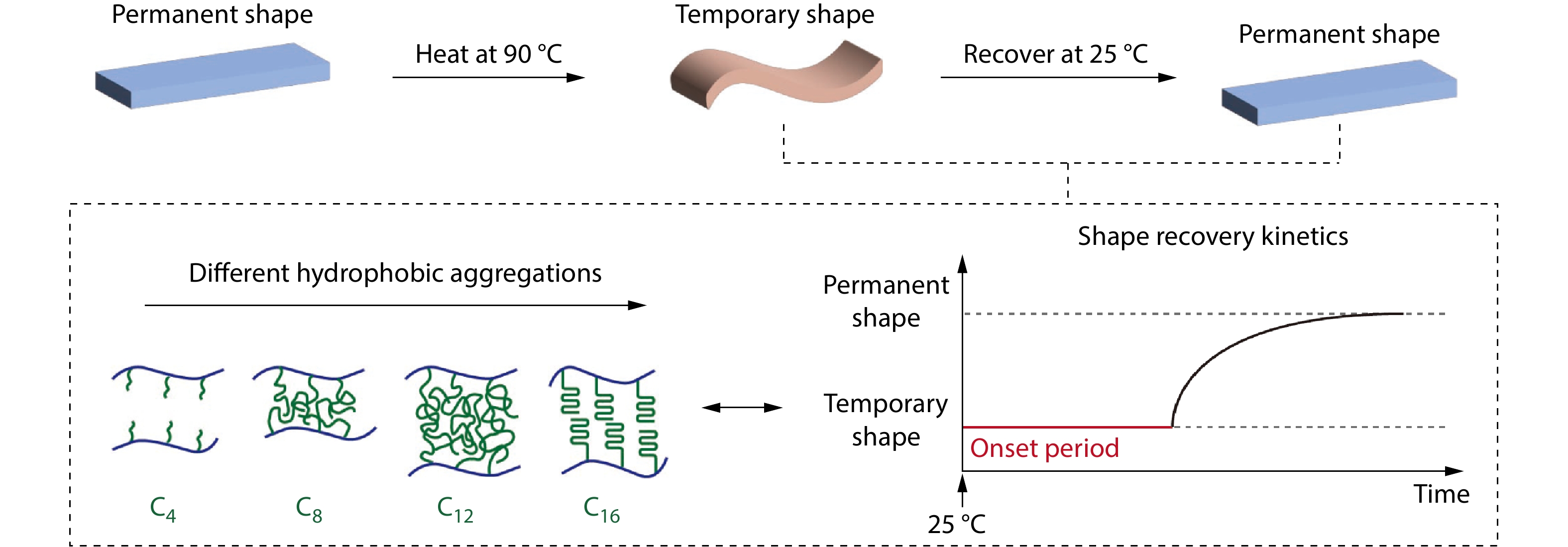
Special Topic: Distinguished Young Scholars in Polymer Science
2026, 44(7): 2051-2061. DOI: 10.1007/s10118-026-3735-1Published(online): 2026-07-01Abstract Full text L-PDFAbstract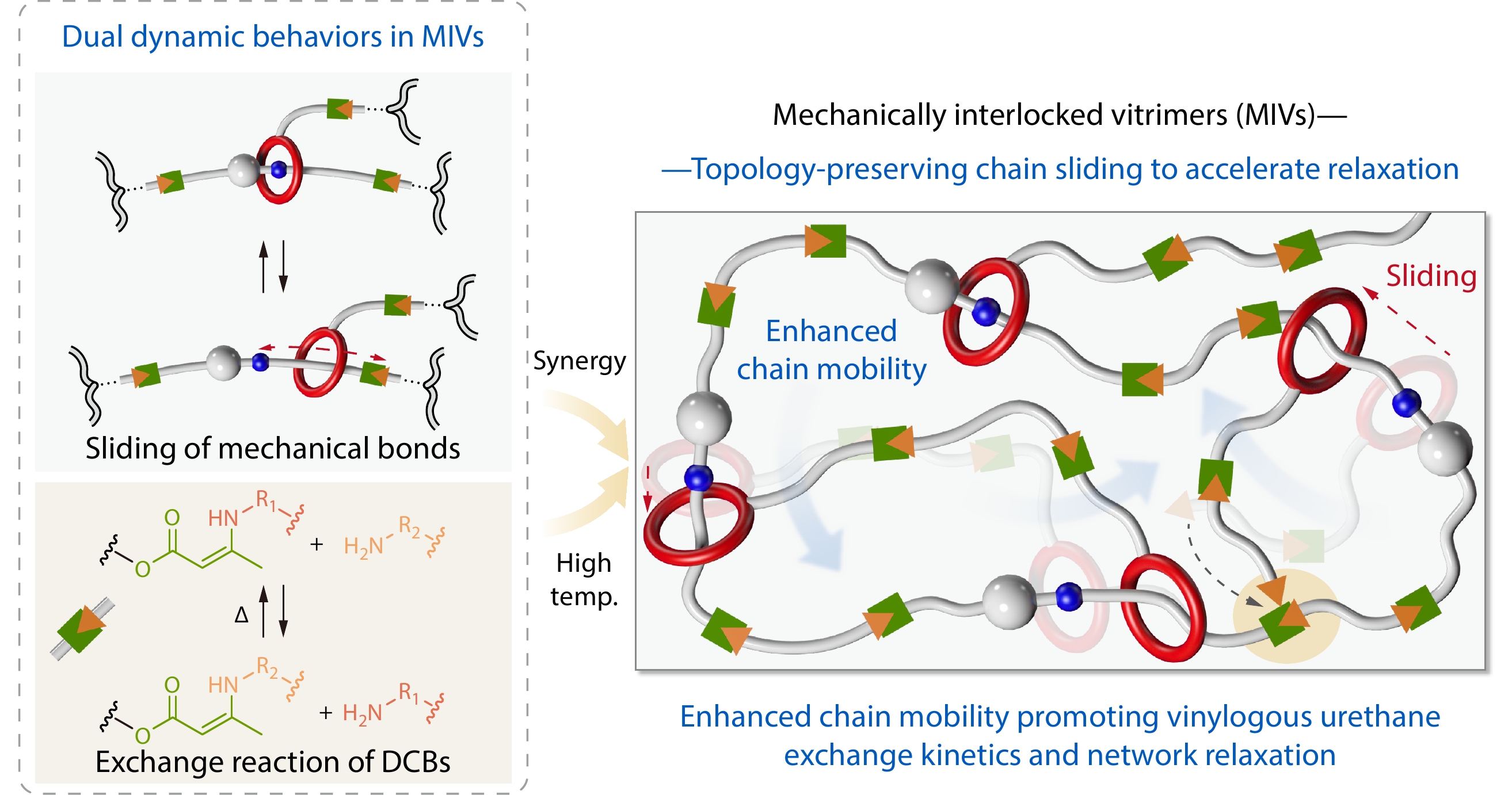

FEATURE ARTICLE
2026, 44(7): 2062-2085. DOI: 10.1007/s10118-026-3621-xPublished(online): 2026-07-01Abstract Full text L-PDFAbstract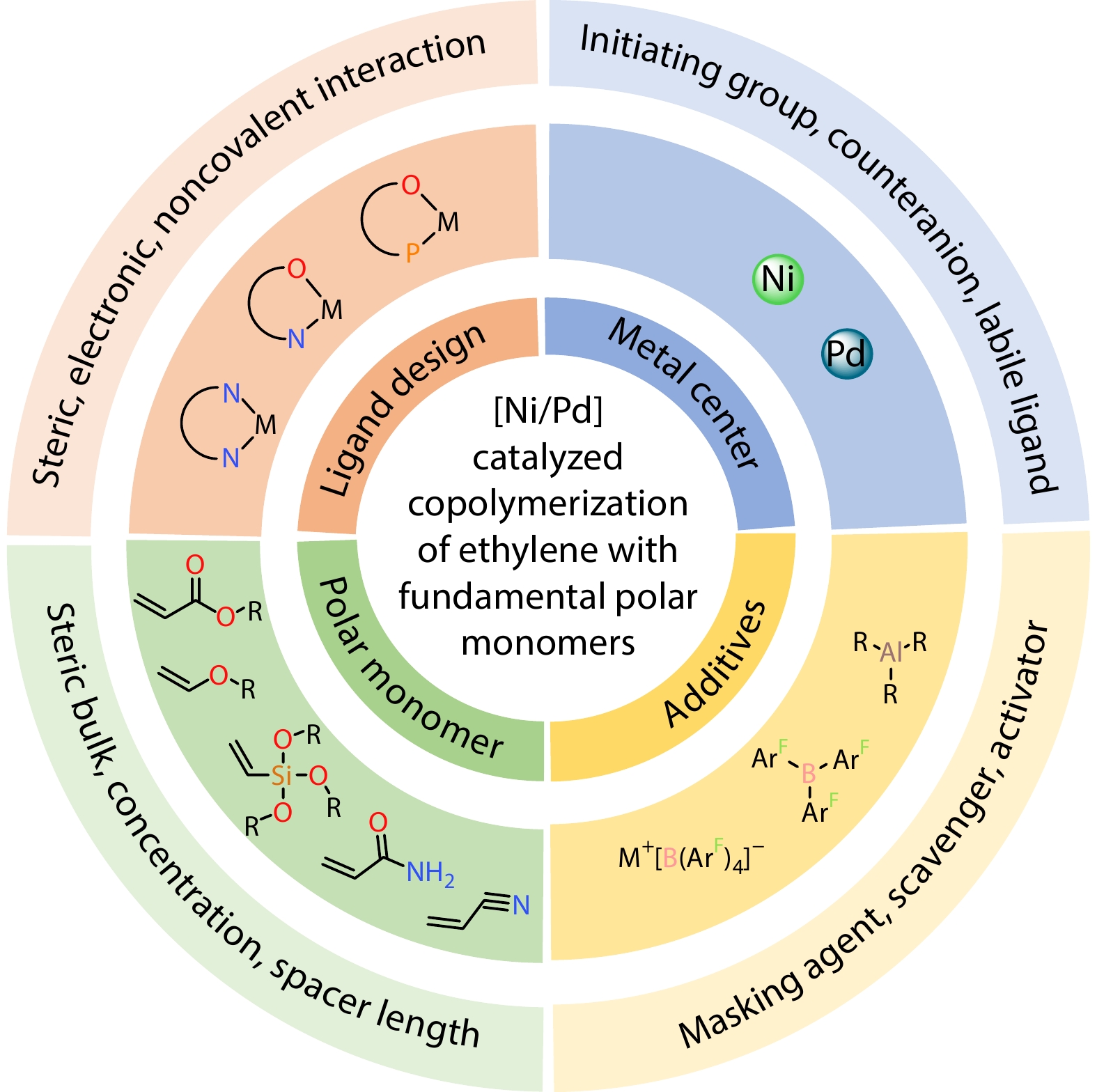
REVIEW
2026, 44(7): 2086-2111. DOI: 10.1007/s10118-026-3635-4Published(online): 2026-07-01Abstract Full text L-PDFAbstract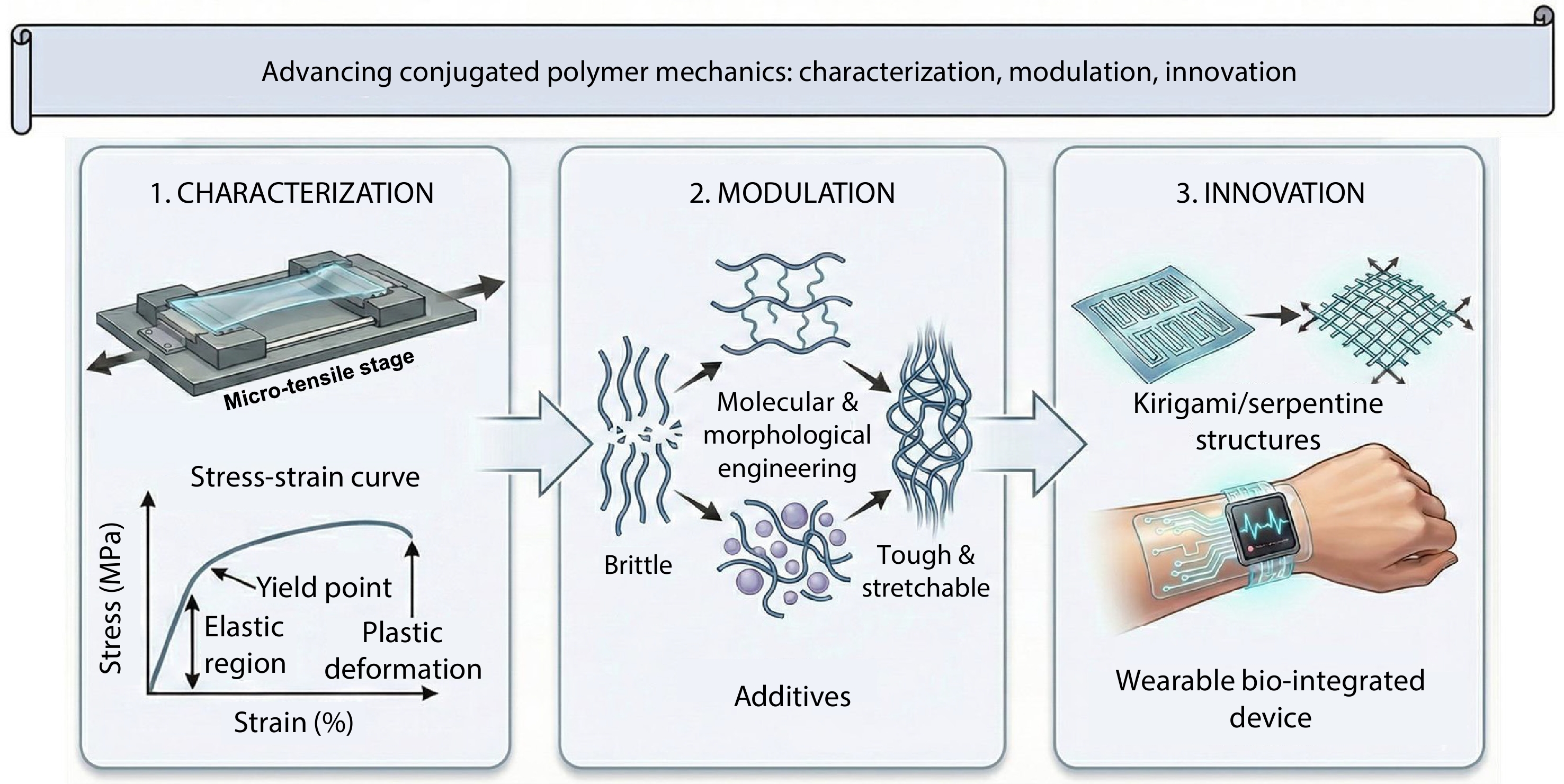
REVIEW
2026, 44(7): 2112-2135. DOI: 10.1007/s10118-026-3633-6Published(online): 2026-07-01Abstract Full text L-PDFAbstract
RESEARCH ARTICLE
2026, 44(7): 2136-2143. DOI: 10.1007/s10118-026-3630-9Published(online): 2026-07-01Abstract Full text L-PDFAbstract
RESEARCH ARTICLE
A Polymer Cathode Interlayer Based on B←N Unit for Suppressed Dark Current in Organic Photodetectors
2026, 44(7): 2144-2152. DOI: 10.1007/s10118-026-3628-3Published(online): 2026-07-01Abstract Full text L-PDFAbstract
RESEARCH ARTICLE
2026, 44(7): 2153-2166. DOI: 10.1007/s10118-026-3600-2Published(online): 2026-07-01Abstract Full text L-PDFAbstract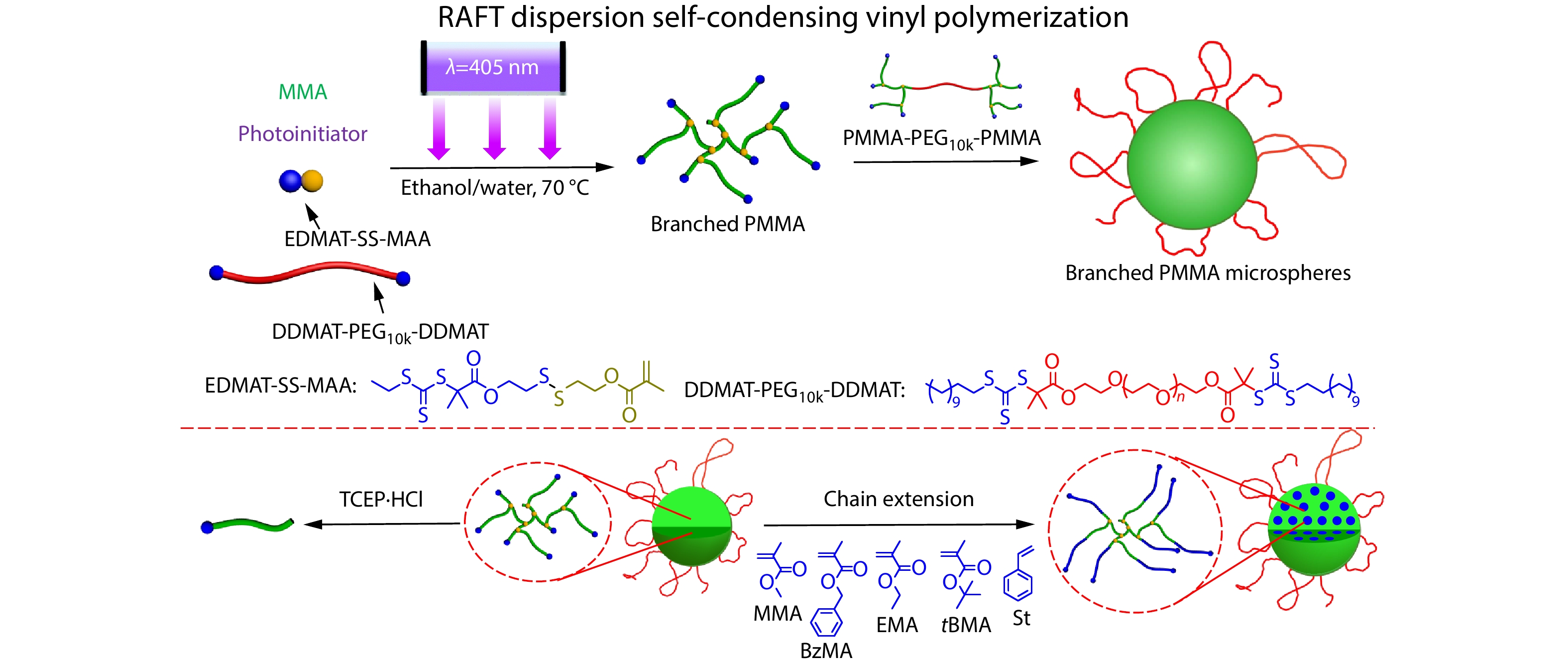
RESEARCH ARTICLE
Microenvironment Responsive Polymeric Nitric Oxide Releasing Micelles for Enhancing Eradication of Methicillin-resistant
Staphylococcus aureus Biofilm Enhanced Publication2026, 44(7): 2167-2176. DOI: 10.1007/s10118-026-3601-1Published(online): 2026-07-01Abstract Full text L-PDFAbstract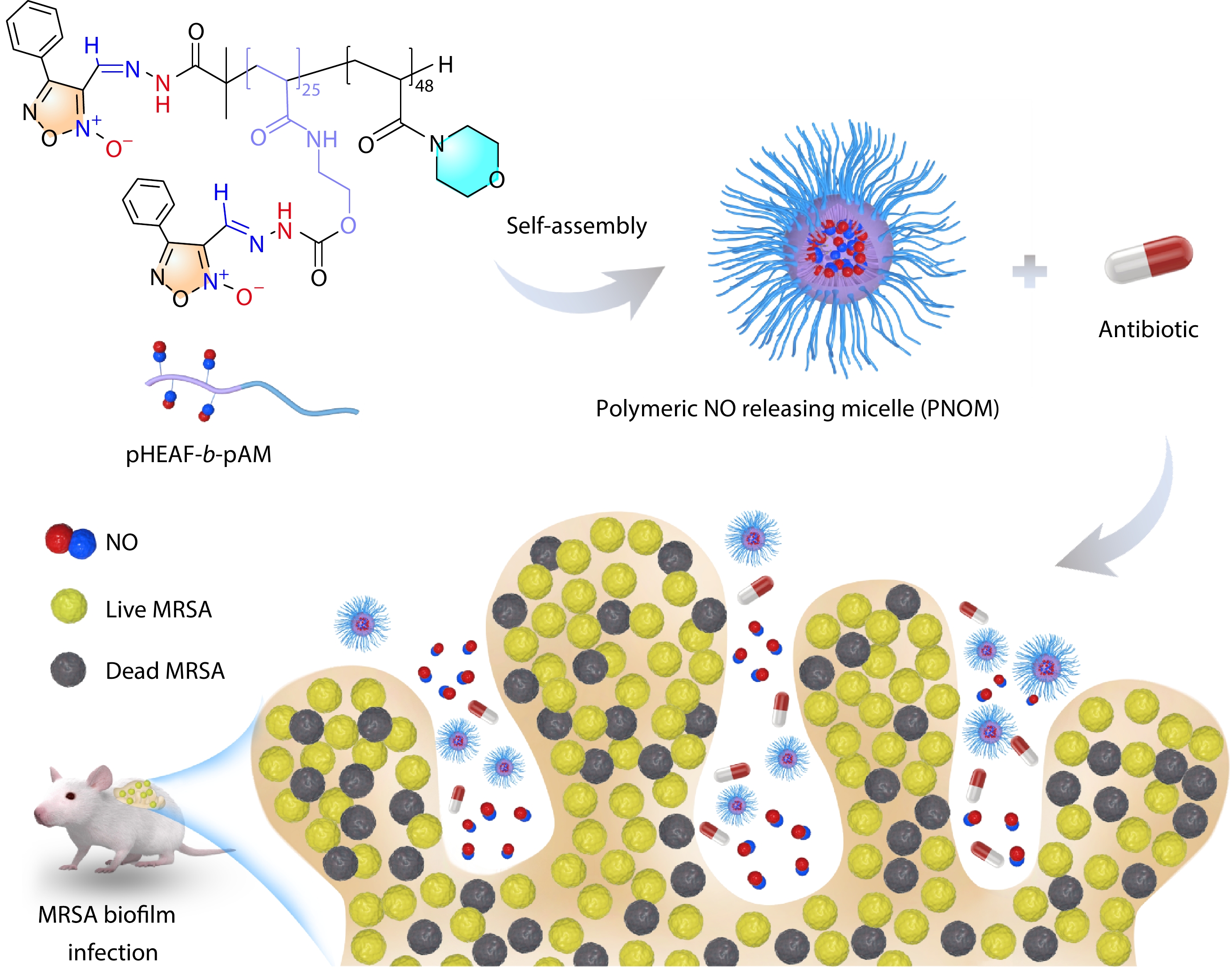
RESEARCH ARTICLE
2026, 44(7): 2177-2187. DOI: 10.1007/s10118-026-3594-9Published(online): 2026-07-01Abstract Full text L-PDFAbstract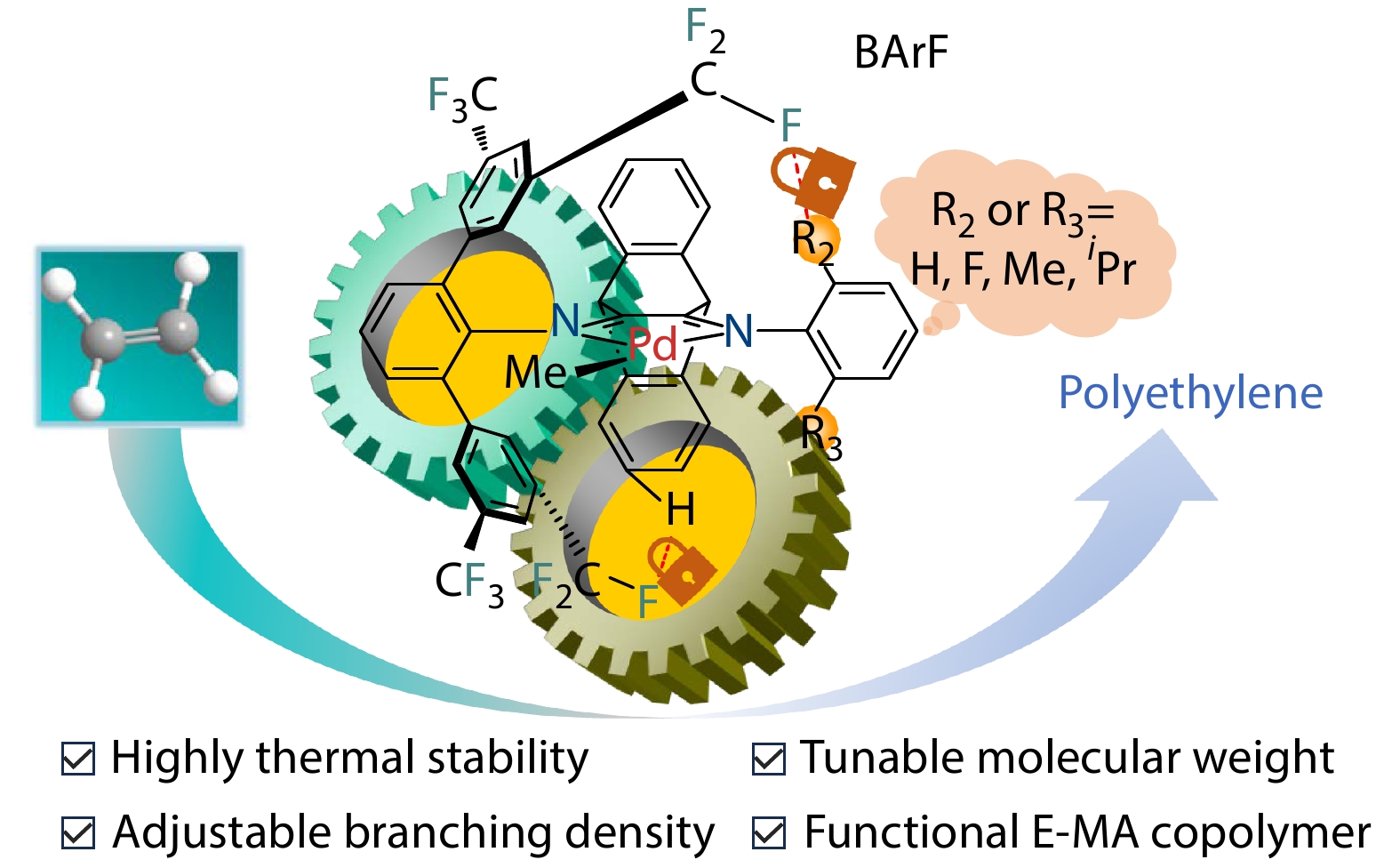
RESEARCH ARTICLE
2026, 44(7): 2188-2198. DOI: 10.1007/s10118-026-3611-zPublished(online): 2026-07-01Abstract Full text L-PDFAbstract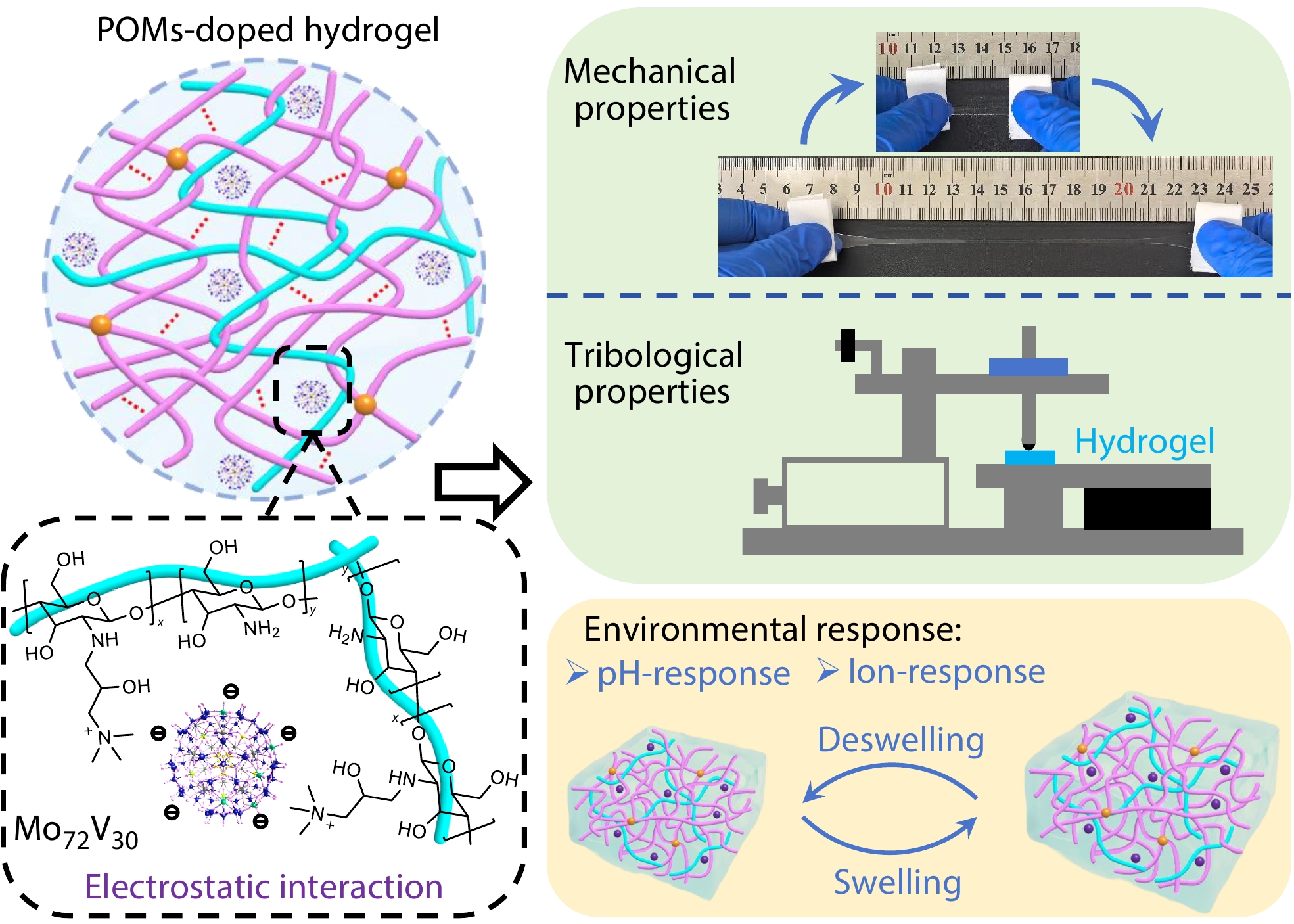
RESEARCH ARTICLE
2026, 44(7): 2199-2211. DOI: 10.1007/s10118-026-3620-yPublished(online): 2026-07-01Abstract Full text L-PDFAbstract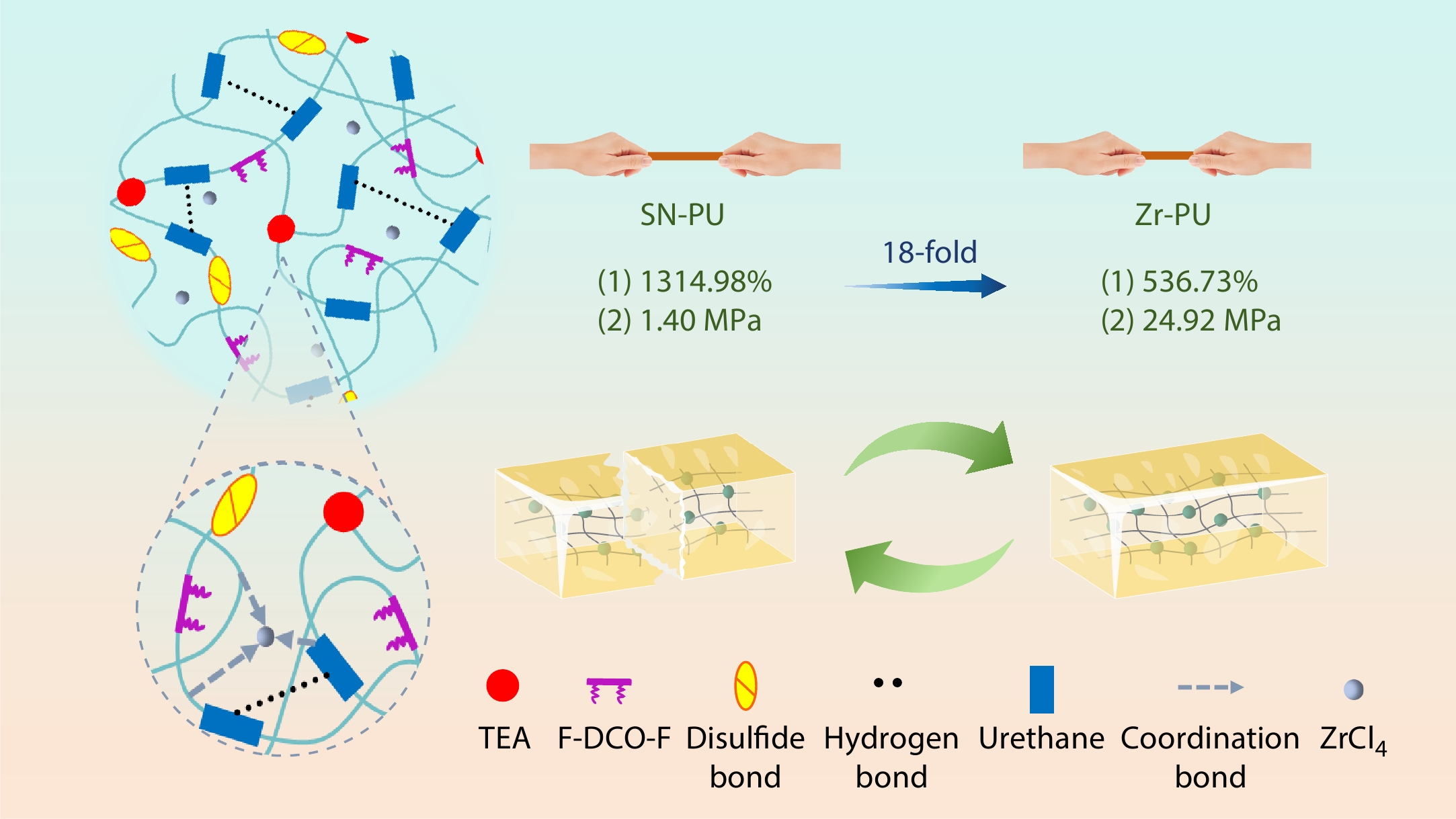
RESEARCH ARTICLE
2026, 44(7): 2212-2225. DOI: 10.1007/s10118-026-3623-8Published(online): 2026-07-01Abstract Full text L-PDFAbstract
RESEARCH ARTICLE
2026, 44(7): 2226-2238. DOI: 10.1007/s10118-026-3624-7Published(online): 2026-07-01Abstract Full text L-PDFAbstract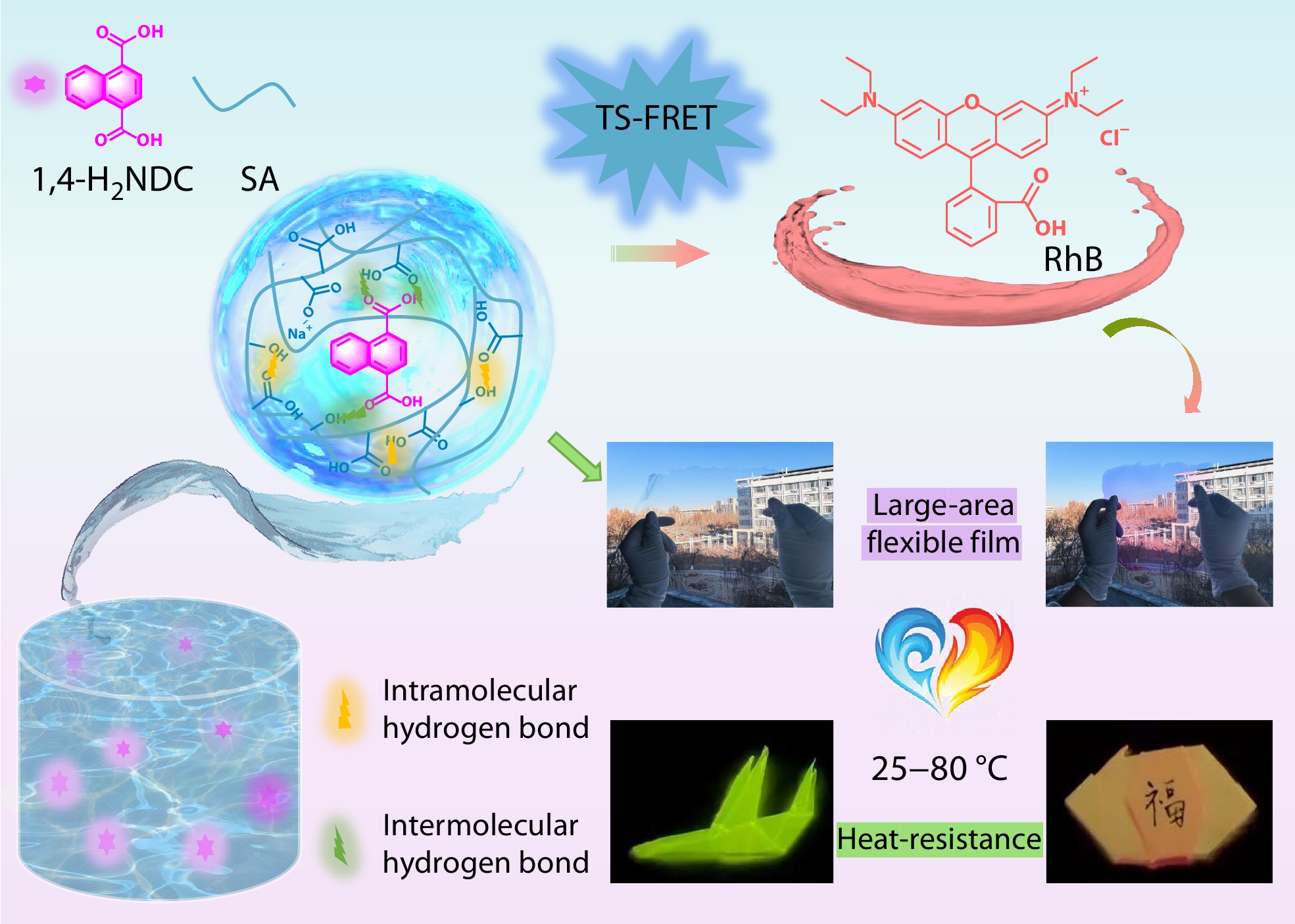
RESEARCH ARTICLE
2026, 44(7): 2239-2247. DOI: 10.1007/s10118-026-3637-2Published(online): 2026-07-01Abstract Full text L-PDFAbstract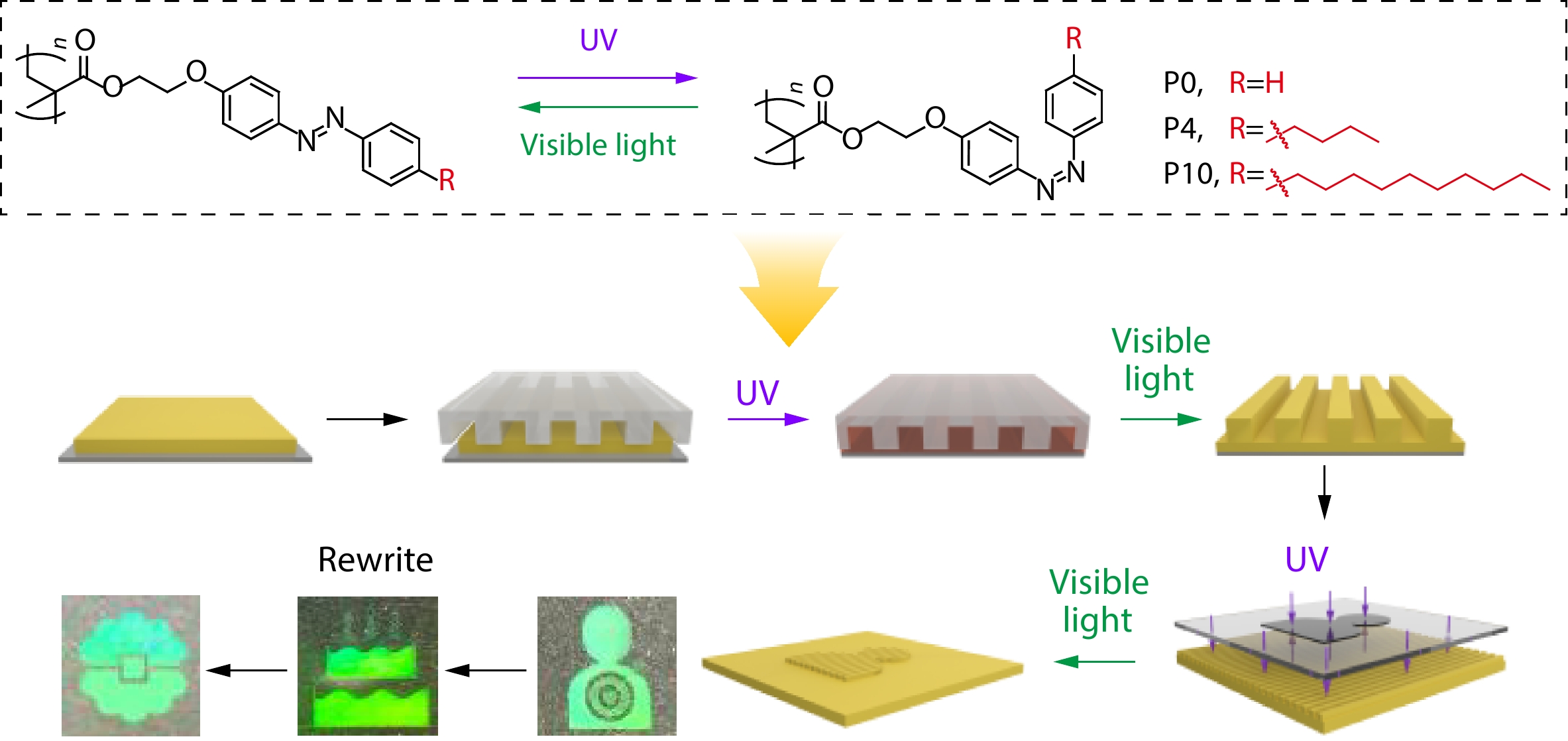
RESEARCH ARTICLE
2026, 44(7): 2248-2257. DOI: 10.1007/s10118-026-3639-0Published(online): 2026-07-01Abstract Full text L-PDFAbstract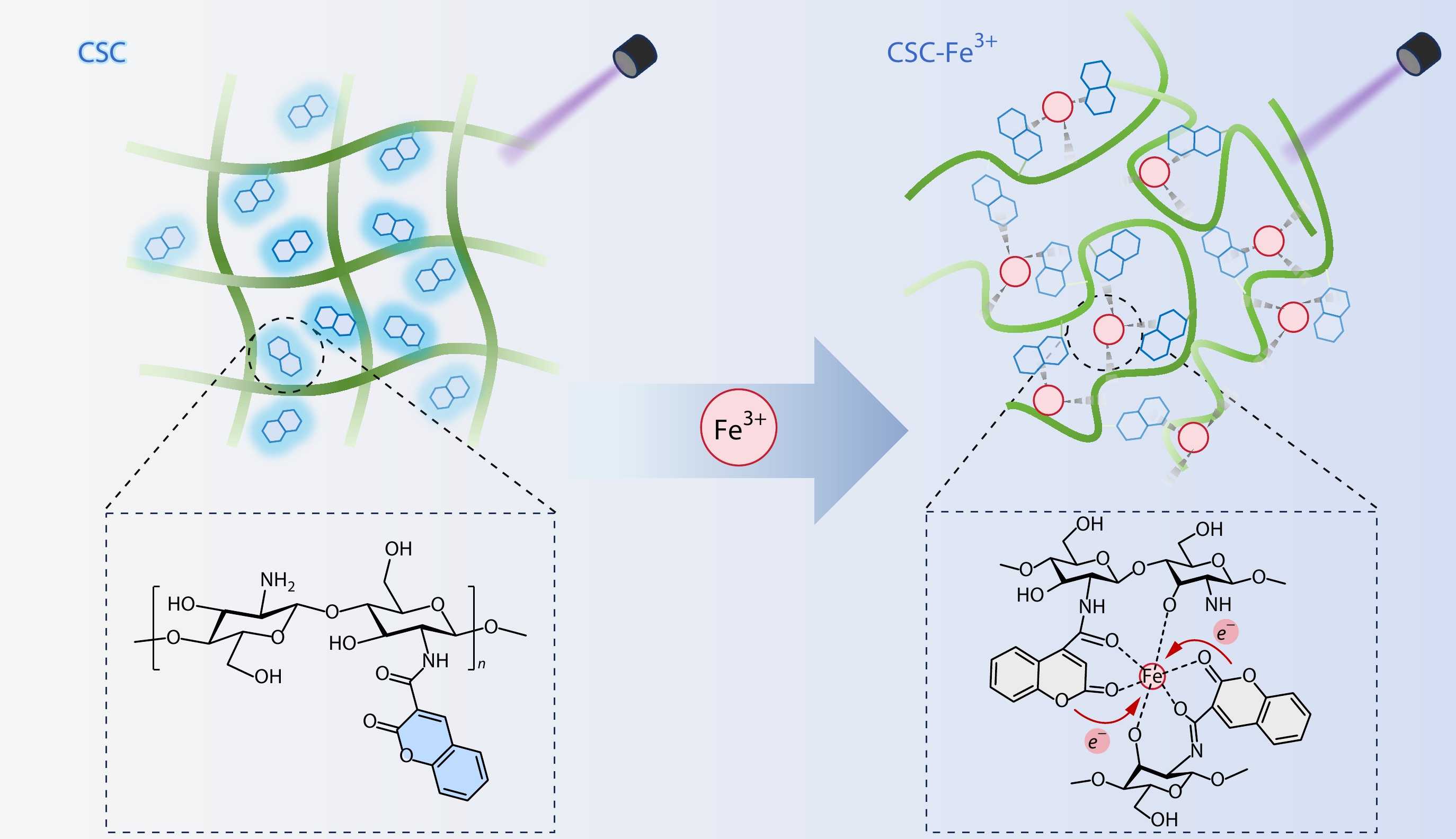
RESEARCH ARTICLE
2026, 44(7): 2258-2268. DOI: 10.1007/s10118-026-3641-6Published(online): 2026-07-01Abstract Full text L-PDFAbstract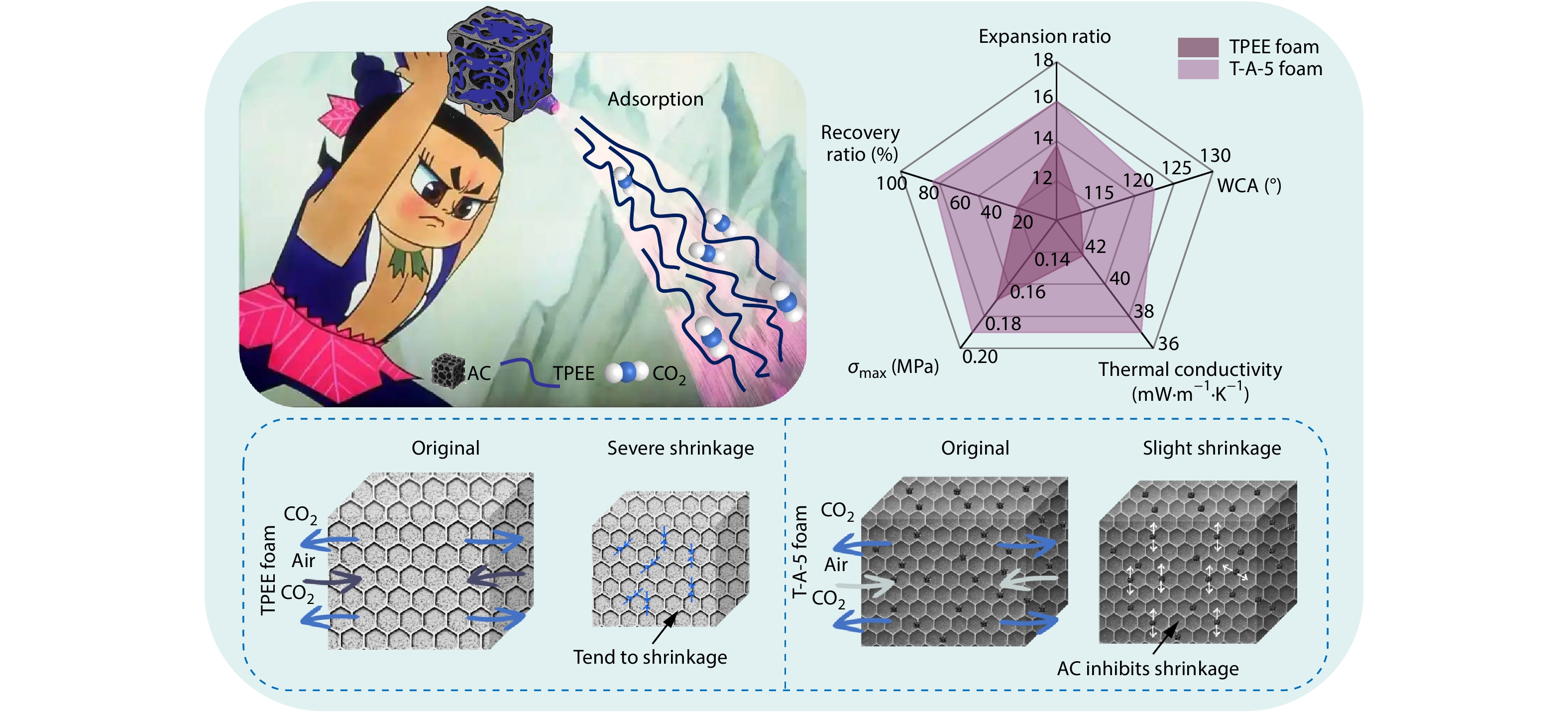
RESEARCH ARTICLE
2026, 44(7): 2269-2281. DOI: 10.1007/s10118-026-3644-3Published(online): 2026-07-01Abstract Full text L-PDFAbstract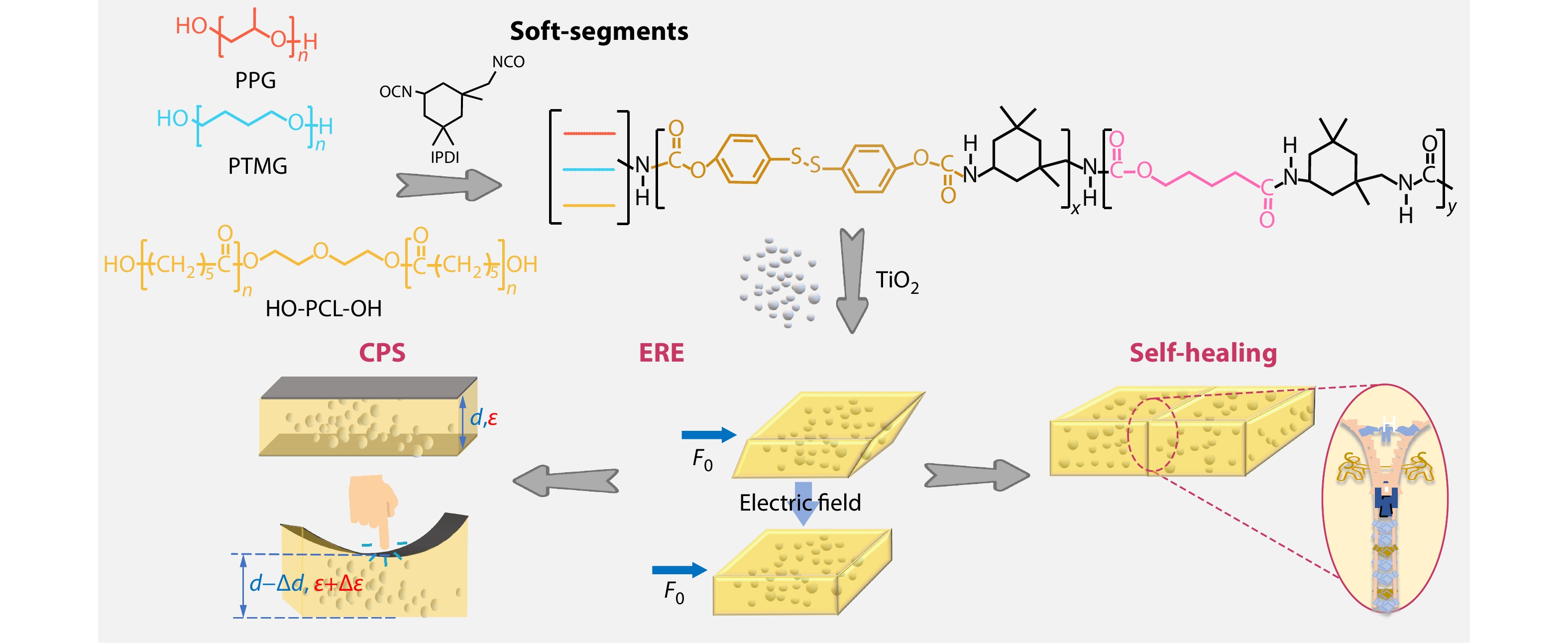
RESEARCH ARTICLE
2026, 44(7): 2282-2293. DOI: 10.1007/s10118-026-3634-5Published(online): 2026-07-01Abstract Full text L-PDFAbstract
RESEARCH ARTICLE
2026, 44(7): 2294-2304. DOI: 10.1007/s10118-026-3588-7Published(online): 2026-07-01Abstract Full text L-PDFAbstract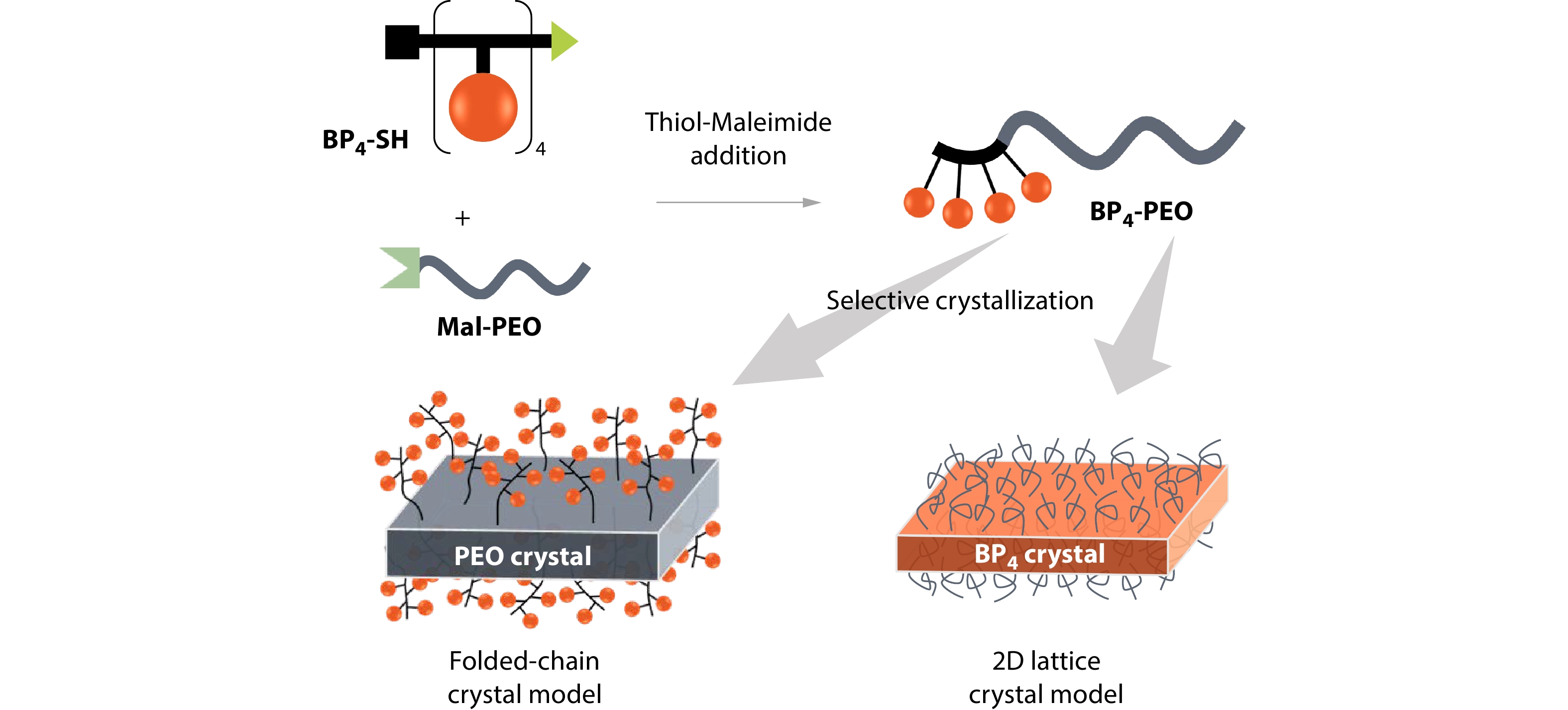
RESEARCH ARTICLE
2026, 44(7): 2305-2321. DOI: 10.1007/s10118-026-3605-xPublished(online): 2026-07-01Abstract Full text L-PDFAbstract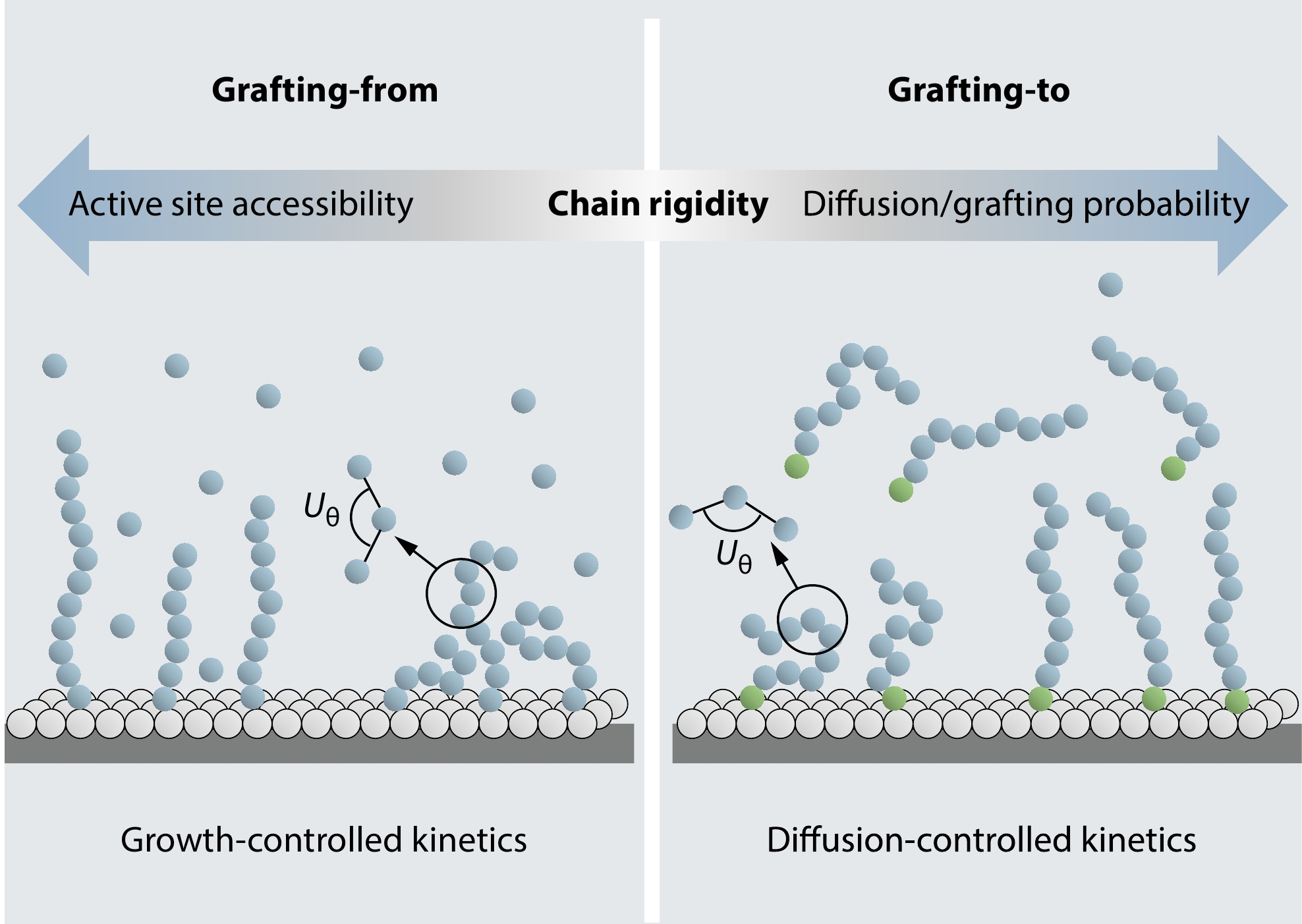
RESEARCH ARTICLE
A Simple Method for Evaluating Periodic Structures Based on Atomic Force Microscopy Images Enhanced Publication
2026, 44(7): 2322-2330. DOI: 10.1007/s10118-026-3625-6Published(online): 2026-07-01Abstract Full text L-PDFAbstract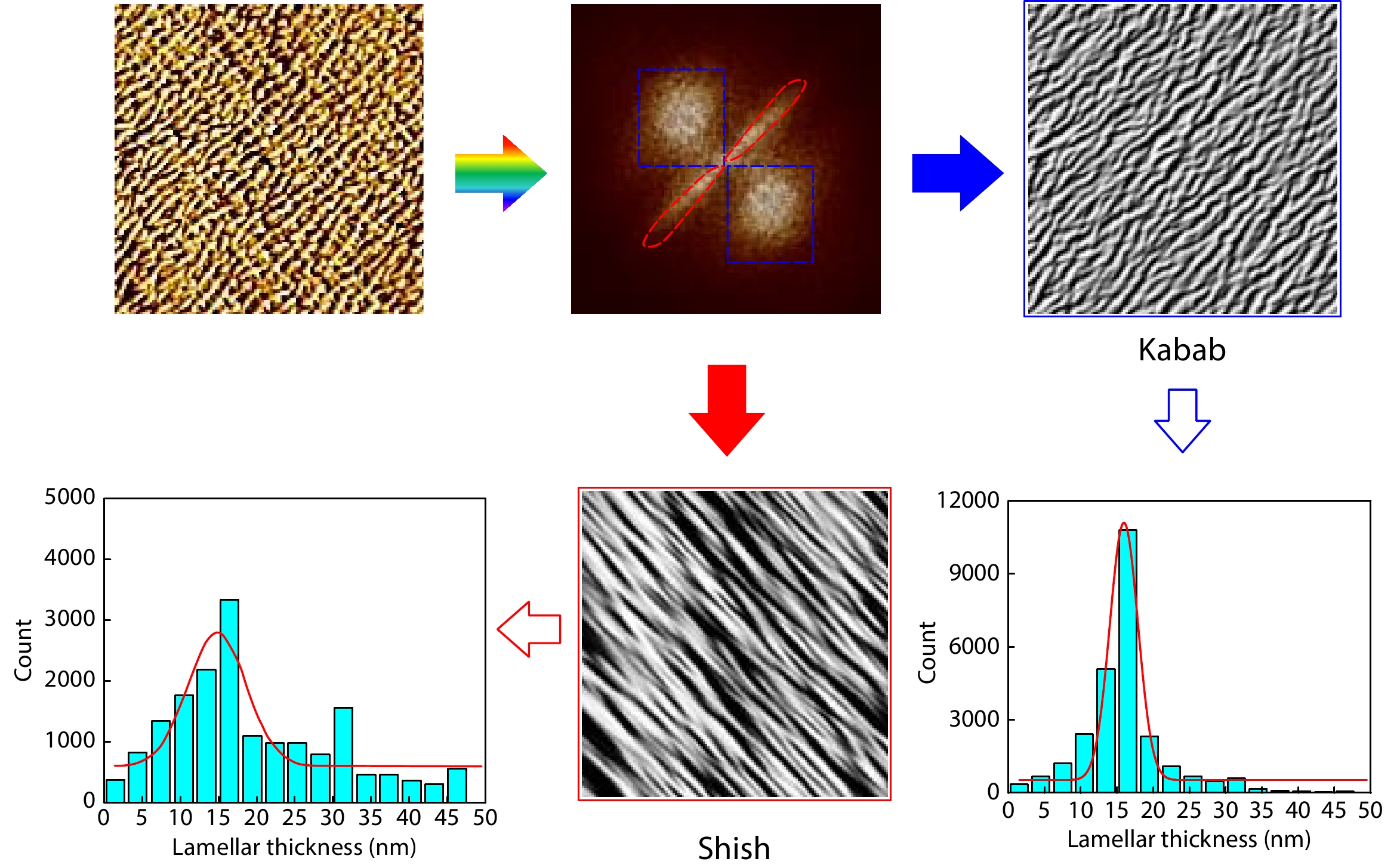
RESEARCH ARTICLE
Molecular Weight Distribution Effects on the Structural and Mechanical Performance of Conjugated Polymers Incorporating Flexible Spacers: A Simulation Study Enhanced Publication
2026, 44(7): 2331-2340. DOI: 10.1007/s10118-026-3626-5Published(online): 2026-07-01Abstract Full text L-PDFAbstract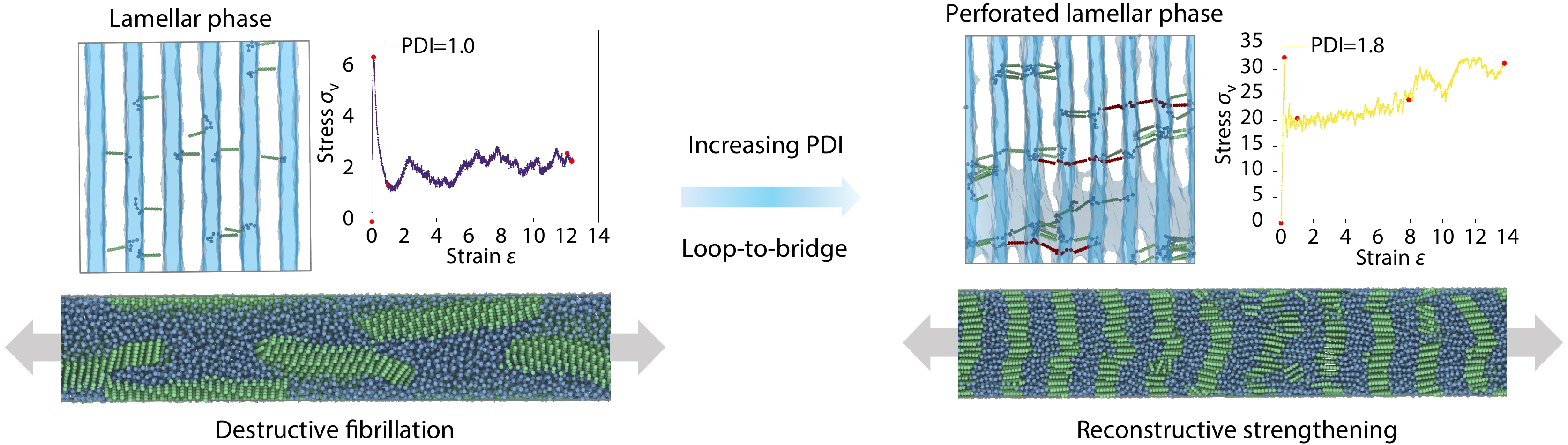
RESEARCH ARTICLE
2026, 44(7): 2341-2351. DOI: 10.1007/s10118-026-3632-7Published(online): 2026-07-01Abstract Full text L-PDFAbstract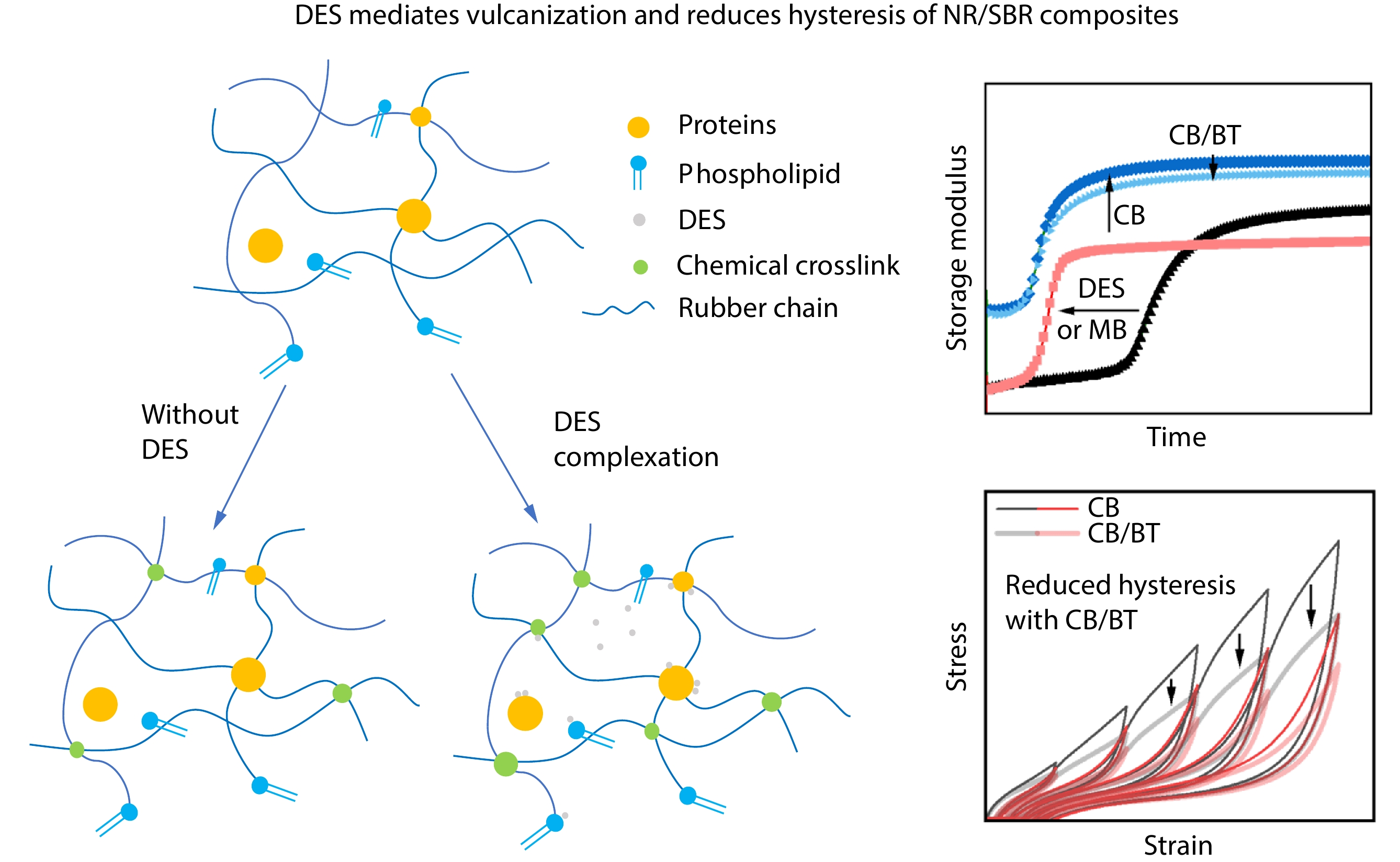
RESEARCH ARTICLE
The Effect of Polymerization of Ionic Liquid [C4Vim]Br on Phase Behavior and Partitioning of L-tryptophan: Experimental and Molecular Dynamics Study Enhanced Publication
2026, 44(7): 2352-2360. DOI: 10.1007/s10118-026-3631-8Published(online): 2026-07-01Abstract Full text L-PDFAbstract![The Effect of Polymerization of Ionic Liquid [C<sub>4</sub>Vim]Br on Phase Behavior and Partitioning of L-tryptophan: Experimental and Molecular Dynamics Study](https://founder-rc-product.oss-cn-zhangjiakou.aliyuncs.com/upload/2026-07-01/112LFQ3O0IAB84424401.jpg?response-content-type=image%2Fjpeg)
RESEARCH ARTICLE
Nanopore-based Manipulation and Separation of Single-stranded DNA Molecules through Tuning pH Values
2026, 44(7): 2361-2369. DOI: 10.1007/s10118-026-3640-7Published(online): 2026-07-01Abstract Full text L-PDFAbstract
RESEARCH ARTICLE
2026, 44(7): 2370-2384. DOI: 10.1007/s10118-026-3657-yPublished(online): 2026-07-01Abstract Full text L-PDFAbstract
RESEARCH ARTICLE
2026, 44(7): 2385-2394. DOI: 10.1007/s10118-026-3627-4Published(online): 2026-07-01Abstract Full text L-PDFAbstract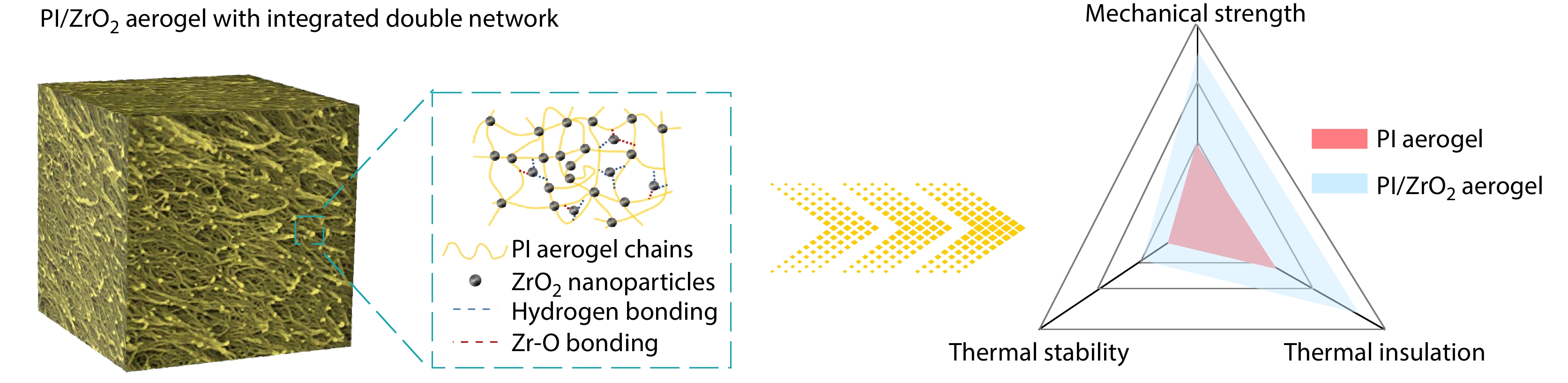
- Technical support is provided by Beijing Founder electronics co., LTD 京ICP备05002797号-9
 京公网安备11010802046900号
京公网安备11010802046900号 - It is recommended to read the content of this site in Chrome&IE9+. Please switch to extreme mode in browser 360.
- Cookies We use cookies to help provide and enhance our service and tailor content. By continuing, you agree to the use of cookies.
0