
Latest Issue
Just Accepted Online First List of Issues Document Types
Issue 3, 2025
RAPID COMMUNICATION
2025, 43(3): 399-405. DOI: 10.1007/s10118-025-3291-0Published(online): 2025-02-25Abstract Full text L-PDFAbstract

REVIEW
2025, 43(3): 406-428. DOI: 10.1007/s10118-025-3279-9Published(online): 2025-02-25Abstract Full text L-PDFAbstract
RESEARCH ARTICLE
2025, 43(3): 429-438. DOI: 10.1007/s10118-025-3275-0Published(online): 2025-02-25Abstract Full text L-PDFAbstract
RESEARCH ARTICLE
2025, 43(3): 439-446. DOI: 10.1007/s10118-025-3277-yPublished(online): 2025-02-25Abstract Full text L-PDFAbstractRESEARCH ARTICLE
2025, 43(3): 447-456. DOI: 10.1007/s10118-025-3290-1Published(online): 2025-02-25Abstract Full text L-PDFAbstract
RESEARCH ARTICLE
2025, 43(3): 457-467. DOI: 10.1007/s10118-025-3273-2Published(online): 2025-02-25Abstract Full text L-PDFAbstract
RESEARCH ARTICLE
2025, 43(3): 468-476. DOI: 10.1007/s10118-025-3282-1Published(online): 2025-02-25Abstract Full text L-PDFAbstract
RESEARCH ARTICLE
2025, 43(3): 477-487. DOI: 10.1007/s10118-025-3296-8Published(online): 2025-02-25Abstract Full text L-PDFAbstract
RESEARCH ARTICLE
2025, 43(3): 488-494. DOI: 10.1007/s10118-025-3289-7Published(online): 2025-02-25Abstract Full text L-PDFAbstract
RESEARCH ARTICLE
2025, 43(3): 495-508. DOI: 10.1007/s10118-025-3287-9Published(online): 2025-02-25Abstract Full text L-PDFAbstract
RESEARCH ARTICLE
2025, 43(3): 509-516. DOI: 10.1007/s10118-025-3268-zPublished(online): 2025-02-25Abstract Full text L-PDFAbstract
RESEARCH ARTICLE
2025, 43(3): 517-532. DOI: 10.1007/s10118-025-3295-9Published(online): 2025-02-25Abstract Full text L-PDFAbstract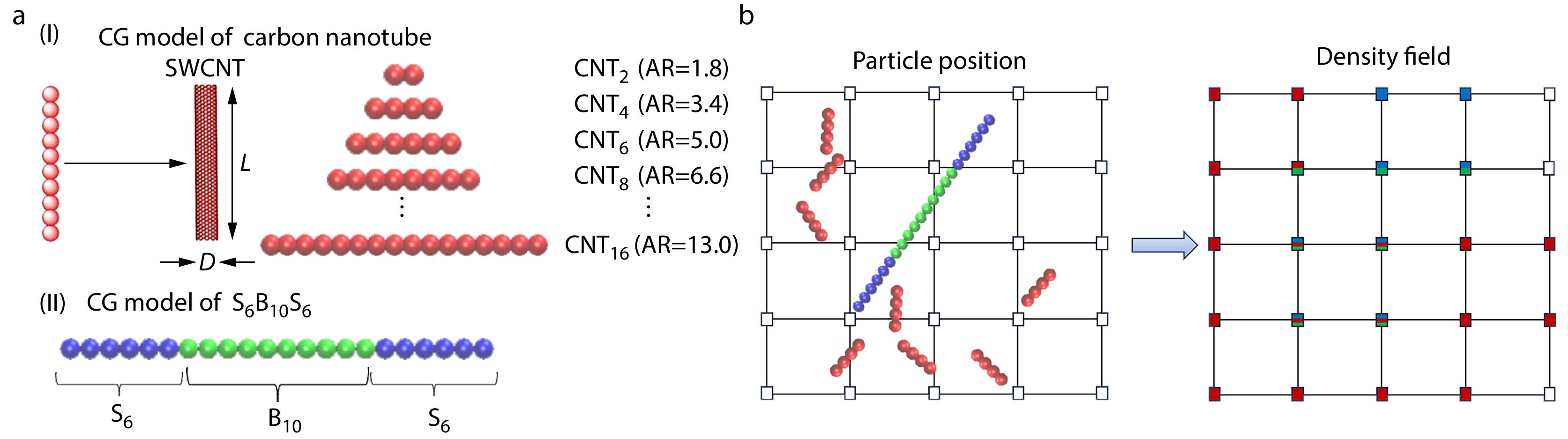
- Technical support is provided by Beijing Founder electronics co., LTD 京ICP备09064830号-19
 京公网安备11010802024621
京公网安备11010802024621 - It is recommended to read the content of this site in Chrome&IE9+. Please switch to extreme mode in browser 360.
- Cookies We use cookies to help provide and enhance our service and tailor content. By continuing, you agree to the use of cookies.
0