Issue Ebook
cover story

Latest Issue
Just Accepted Online First List of Issues Document Types
Issue 4, 2026
PREFACE
2026, 44(4): 905. DOI: 10.1007/s10118-026-3649-yPublished(online): 2026-03-26Full text L-PDF

REVIEW
2026, 44(4): 906-921. DOI: 10.1007/s10118-026-3622-9Published(online): 2026-03-26Abstract Full text L-PDFAbstract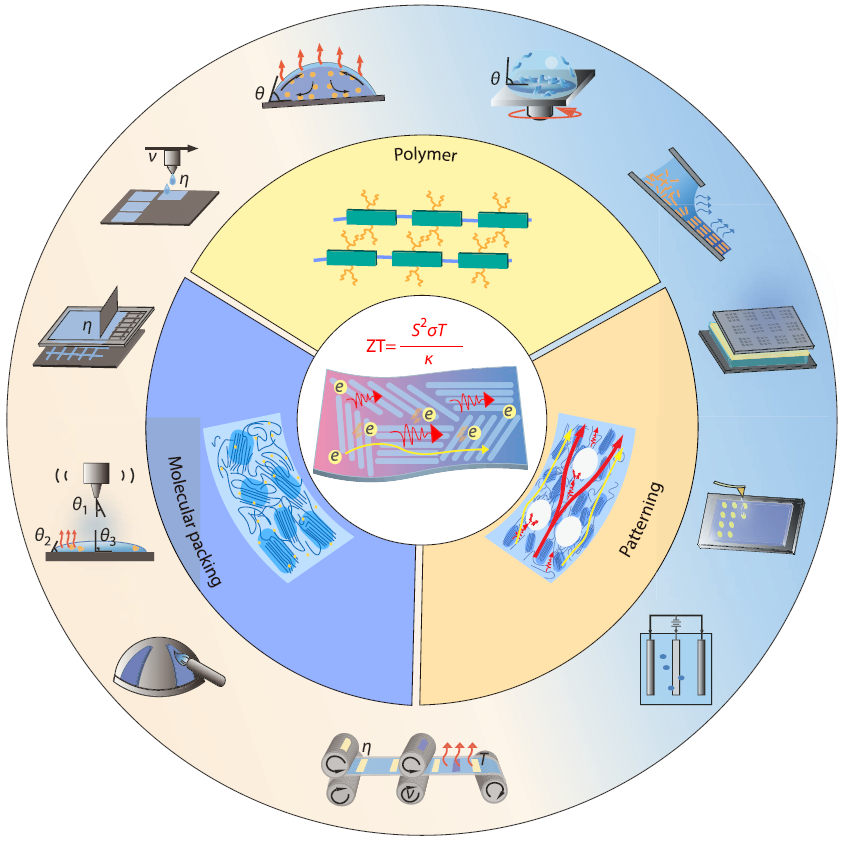
RESEARCH ARTICLE
Erosion-immune Layer-by-layer Deposition Enabled by Interfacial Buffering toward 20.21%-Efficient Pseudo-Planar Heterojunction Organic Solar Cells Enhanced Publication
2026, 44(4): 922-936. DOI: 10.1007/s10118-025-3500-xPublished(online): 2026-03-26Abstract Full text L-PDFAbstract
RESEARCH ARTICLE
2026, 44(4): 937-949. DOI: 10.1007/s10118-025-3496-2Published(online): 2026-03-26Abstract Full text L-PDFAbstract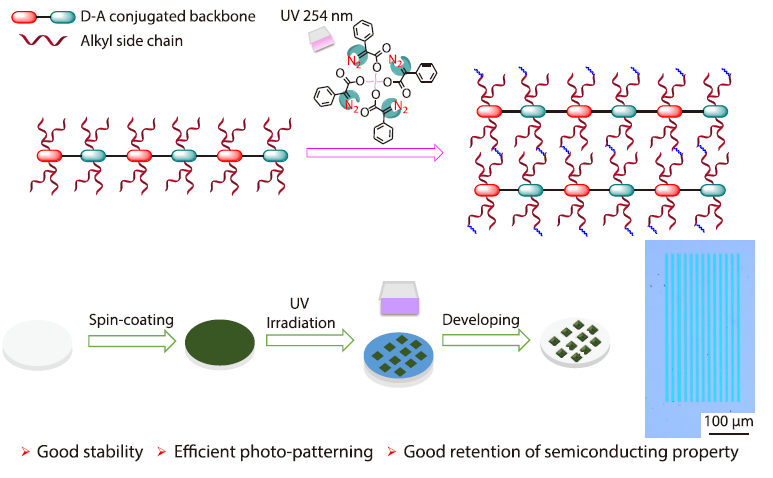
RESEARCH ARTICLE
2026, 44(4): 950-958. DOI: 10.1007/s10118-025-3512-6Published(online): 2026-03-26Abstract Full text L-PDFAbstract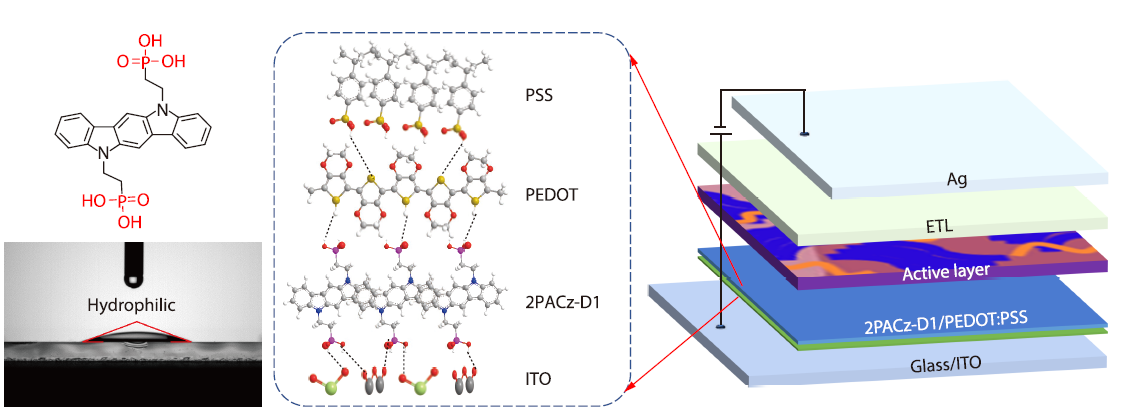
RESEARCH ARTICLE
Plasticizer Enhanced Chain Orientation and Dynamics for Printed Stretchable Conjugated Polymer Films
2026, 44(4): 959-969. DOI: 10.1007/s10118-025-3526-0Published(online): 2026-03-26Abstract Full text L-PDFAbstract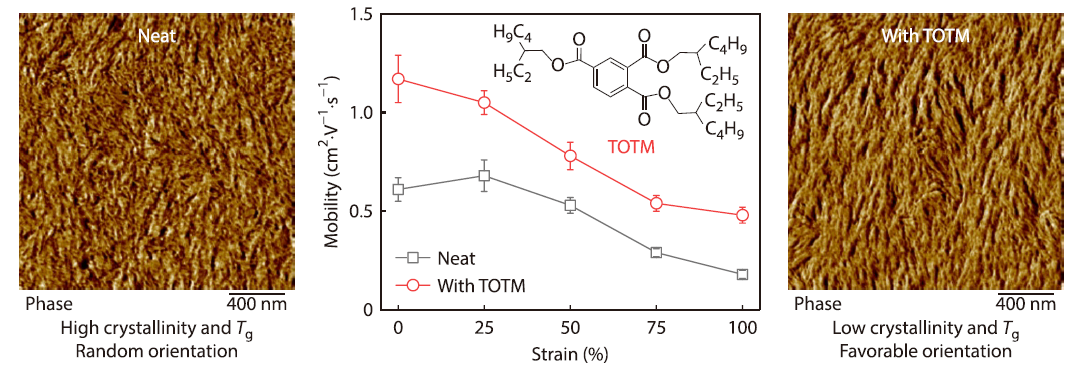
RESEARCH ARTICLE
2026, 44(4): 970-979. DOI: 10.1007/s10118-025-3525-1Published(online): 2026-03-26Abstract Full text L-PDFAbstract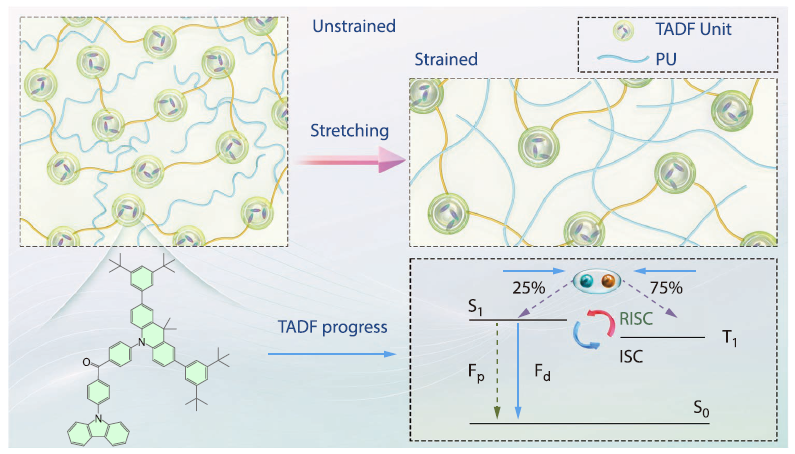
RESEARCH ARTICLE
Meta-linked Phenoselenazine-based Conjugated Polymers for Efficient Room-temperature Phosphorescence
2026, 44(4): 980-987. DOI: 10.1007/s10118-025-3517-1Published(online): 2026-03-26Abstract Full text L-PDFAbstract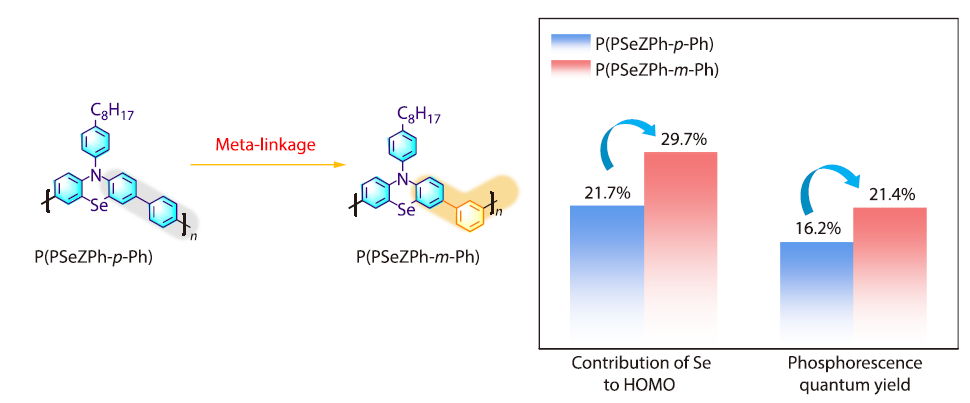
RESEARCH ARTICLE
2026, 44(4): 988-995. DOI: 10.1007/s10118-026-3559-zPublished(online): 2026-03-26Abstract Full text L-PDFAbstract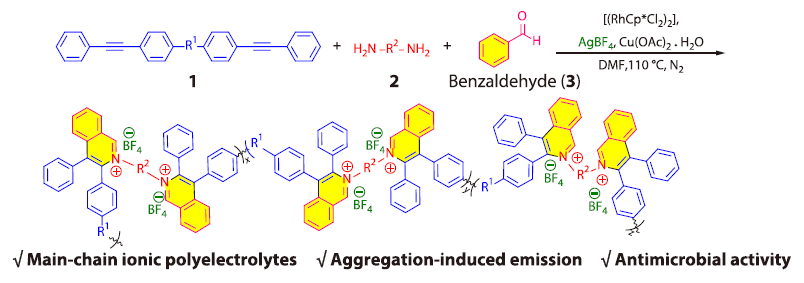
RESEARCH ARTICLE
2026, 44(4): 996-1006. DOI: 10.1007/s10118-026-3566-0Published(online): 2026-03-26Abstract Full text L-PDFAbstract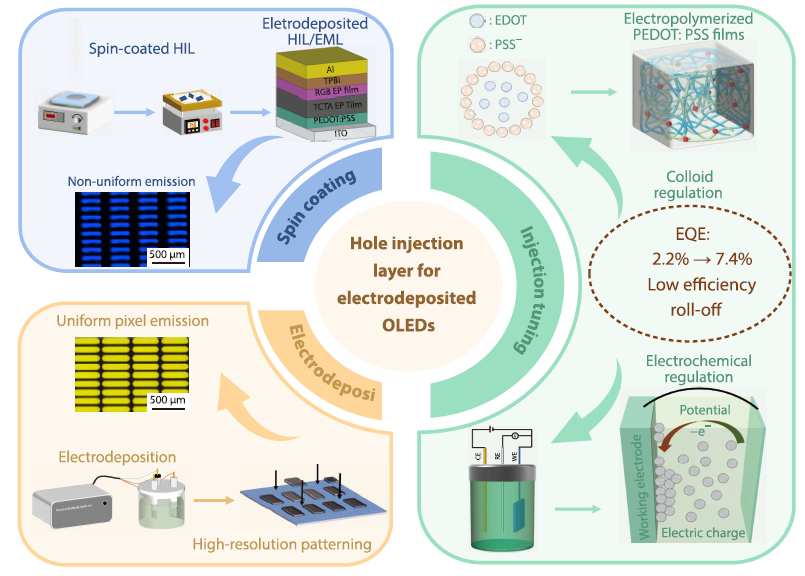
RESEARCH ARTICLE
2026, 44(4): 1007-1016. DOI: 10.1007/s10118-026-3579-8Published(online): 2026-03-26Abstract Full text L-PDFAbstract
RESEARCH ARTICLE
2026, 44(4): 1017-1026. DOI: 10.1007/s10118-026-3560-6Published(online): 2026-03-26Abstract Full text L-PDFAbstract
RESEARCH ARTICLE
2026, 44(4): 1027-1034. DOI: 10.1007/s10118-026-3572-2Published(online): 2026-03-26Abstract Full text L-PDFAbstract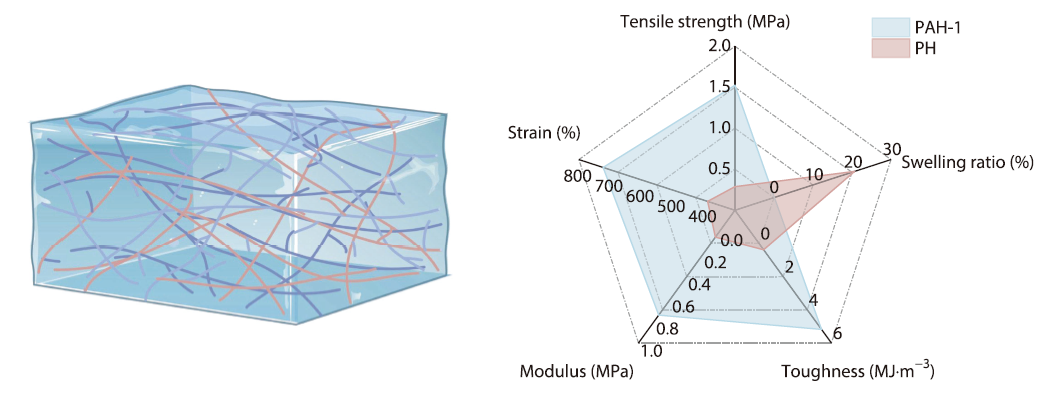
RESEARCH ARTICLE
2026, 44(4): 1035-1045. DOI: 10.1007/s10118-026-3555-3Published(online): 2026-03-26Abstract Full text L-PDFAbstract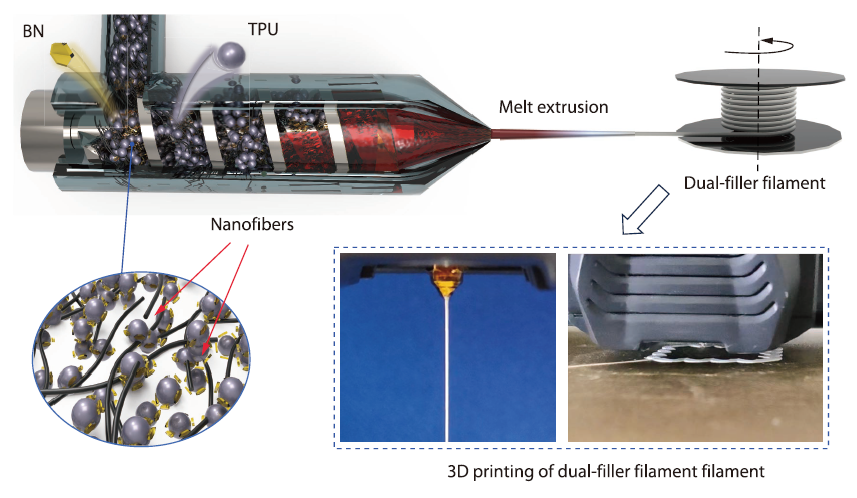
RESEARCH ARTICLE
2026, 44(4): 1046-1058. DOI: 10.1007/s10118-026-3554-4Published(online): 2026-03-26Abstract Full text L-PDFAbstract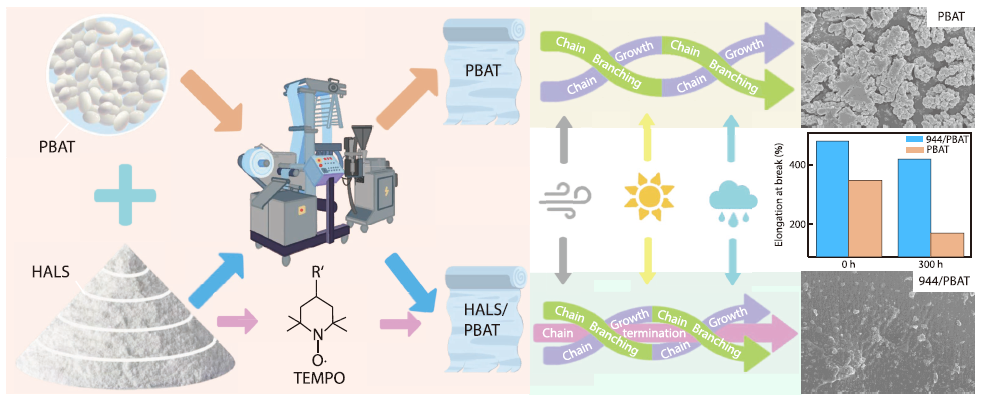
RESEARCH ARTICLE
2026, 44(4): 1059-1068. DOI: 10.1007/s10118-026-3562-4Published(online): 2026-03-26Abstract Full text L-PDFAbstract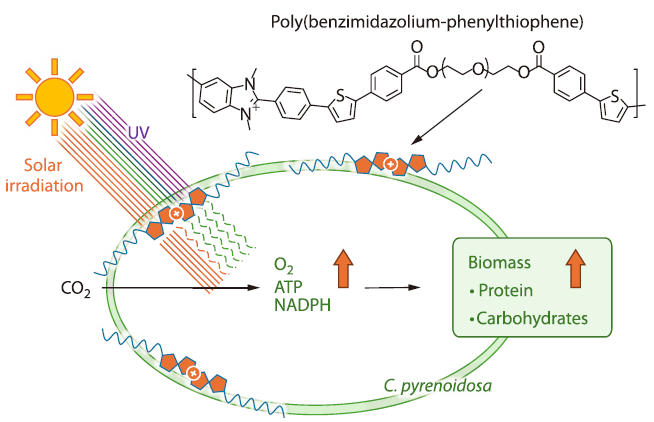
RESEARCH ARTICLE
2026, 44(4): 1069-1082. DOI: 10.1007/s10118-026-3561-5Published(online): 2026-03-26Abstract Full text L-PDFAbstract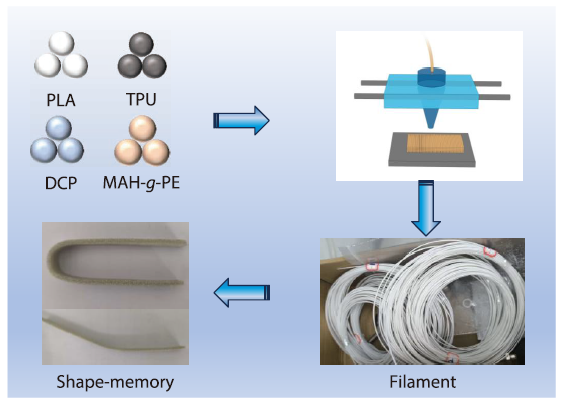
RESEARCH ARTICLE
2026, 44(4): 1083-1089. DOI: 10.1007/s10118-026-3577-xPublished(online): 2026-03-26Abstract Full text L-PDFAbstract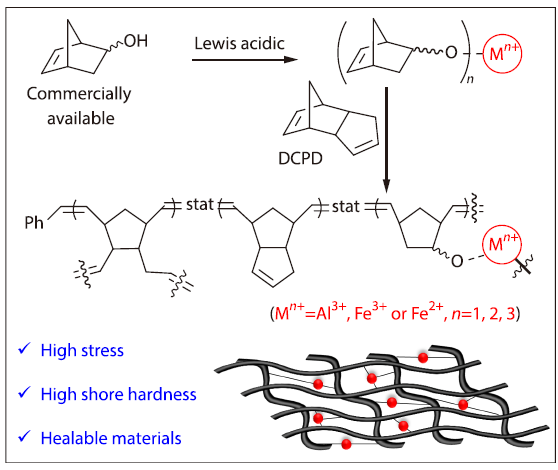
RESEARCH ARTICLE
2026, 44(4): 1090-1101. DOI: 10.1007/s10118-026-3574-0Published(online): 2026-03-26Abstract Full text L-PDFAbstract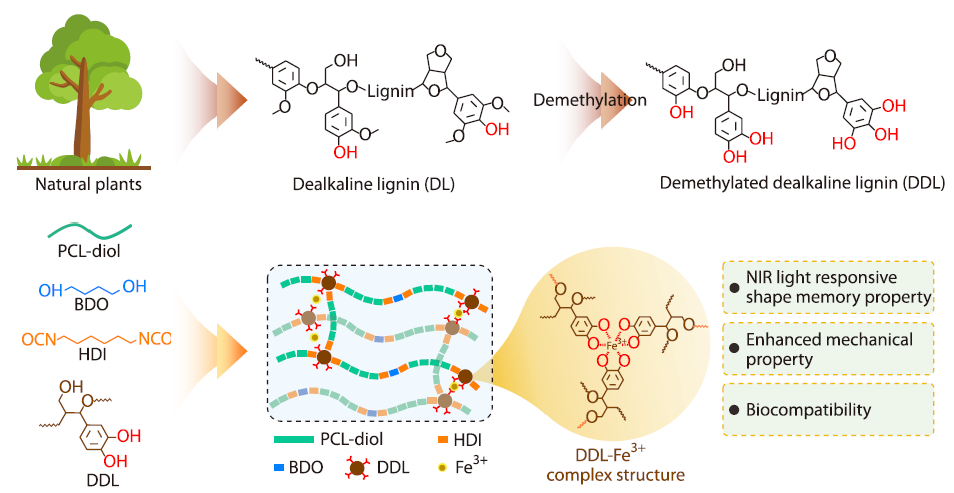
RESEARCH ARTICLE
2026, 44(4): 1102-1113. DOI: 10.1007/s10118-026-3557-1Published(online): 2026-03-26Abstract Full text L-PDFAbstract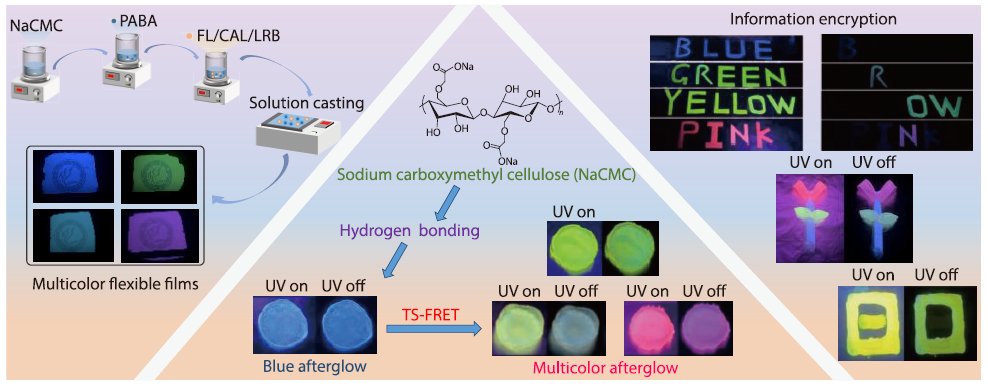
RESEARCH ARTICLE
2026, 44(4): 1114-1125. DOI: 10.1007/s10118-026-3558-0Published(online): 2026-03-26Abstract Full text L-PDFAbstract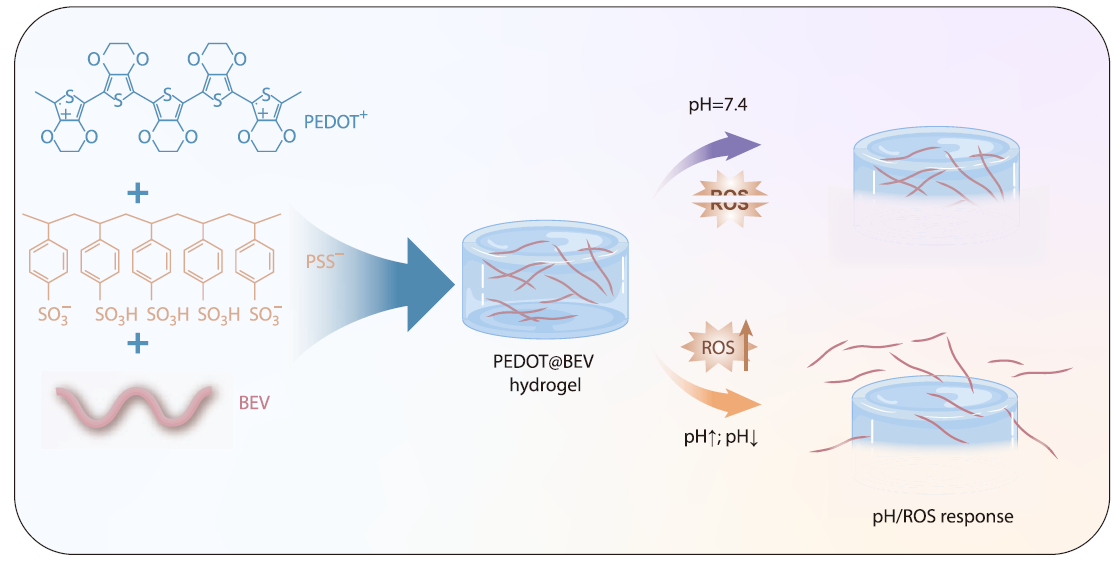
RESEARCH ARTICLE
2026, 44(4): 1126-1141. DOI: 10.1007/s10118-026-3569-xPublished(online): 2026-03-26Abstract Full text L-PDFAbstract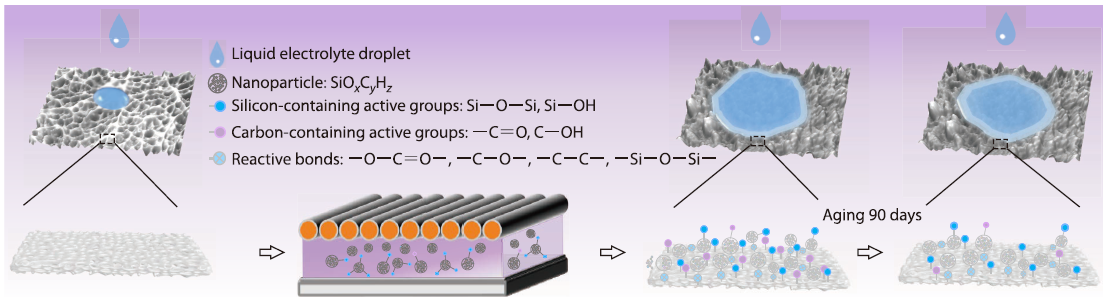
RESEARCH ARTICLE
2026, 44(4): 1142-1153. DOI: 10.1007/s10118-026-3565-1Published(online): 2026-03-26Abstract Full text L-PDFAbstract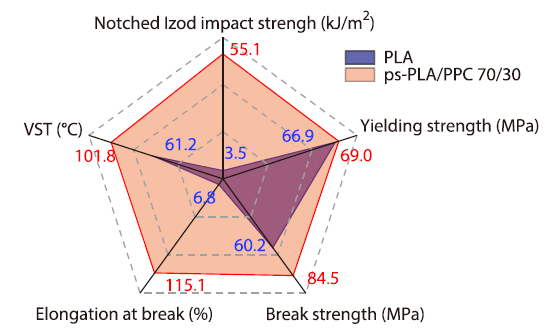
RESEARCH ARTICLE
2026, 44(4): 1154-1164. DOI: 10.1007/s10118-026-3563-3Published(online): 2026-03-26Abstract Full text L-PDFAbstract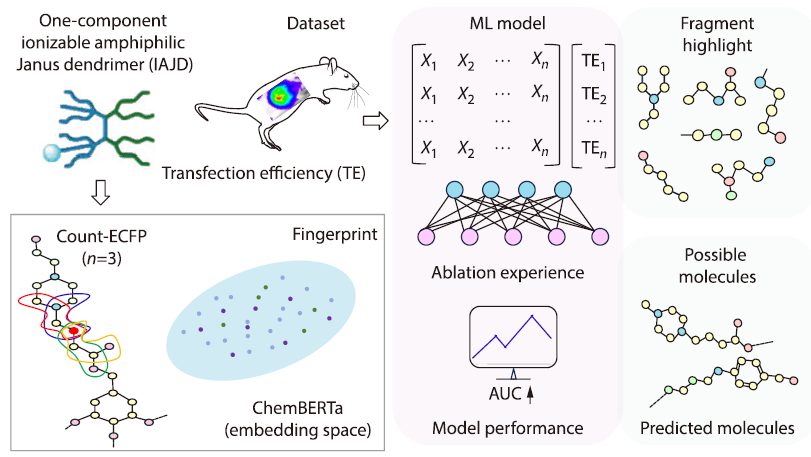
RESEARCH ARTICLE
2026, 44(4): 1165-1172. DOI: 10.1007/s10118-026-3568-yPublished(online): 2026-03-26Abstract Full text L-PDFAbstract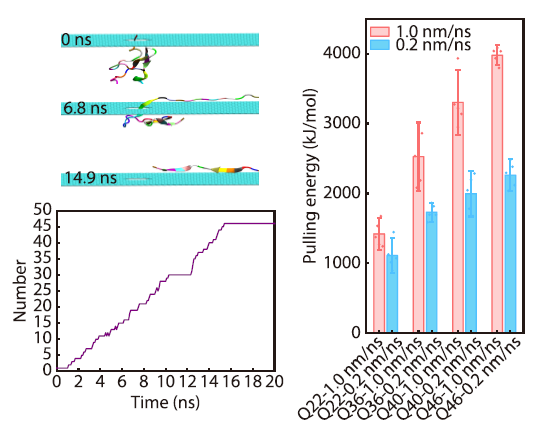
RESEARCH ARTICLE
2026, 44(4): 1173-1185. DOI: 10.1007/s10118-026-3575-zPublished(online): 2026-03-26Abstract Full text L-PDFAbstract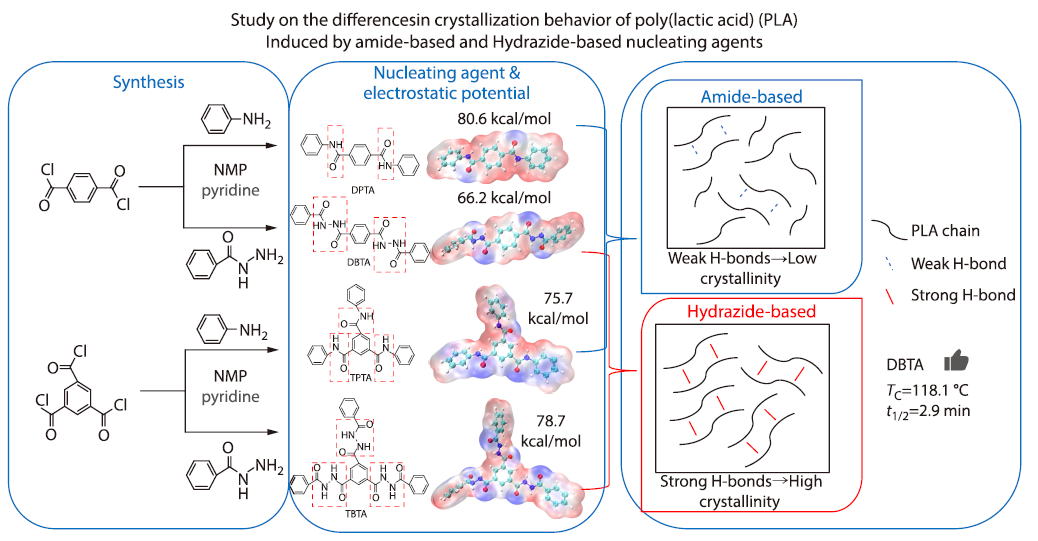
RESEARCH ARTICLE
2026, 44(4): 1186-1198. DOI: 10.1007/s10118-026-3580-2Published(online): 2026-03-26Abstract Full text L-PDFAbstract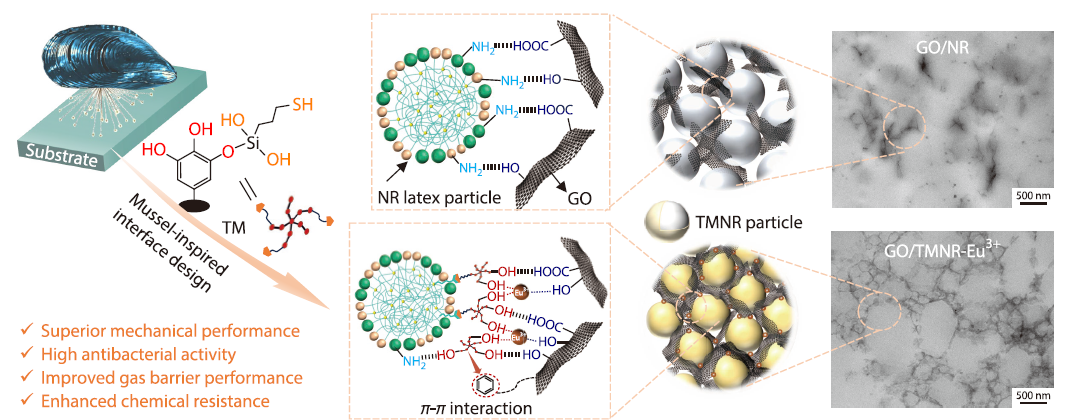
- Technical support is provided by Beijing Founder electronics co., LTD 京ICP备05002797号-9
 京公网安备11010802046900号
京公网安备11010802046900号 - It is recommended to read the content of this site in Chrome&IE9+. Please switch to extreme mode in browser 360.
- Cookies We use cookies to help provide and enhance our service and tailor content. By continuing, you agree to the use of cookies.
0