
Latest Issue
Just Accepted Online First List of Issues Document Types
Issue 9, 2025
RESEARCH ARTICLE
2025, 43(9): 1483-1495. DOI: 10.1007/s10118-025-3388-5Published(online): 2025-08-25Abstract Full text L-PDFAbstract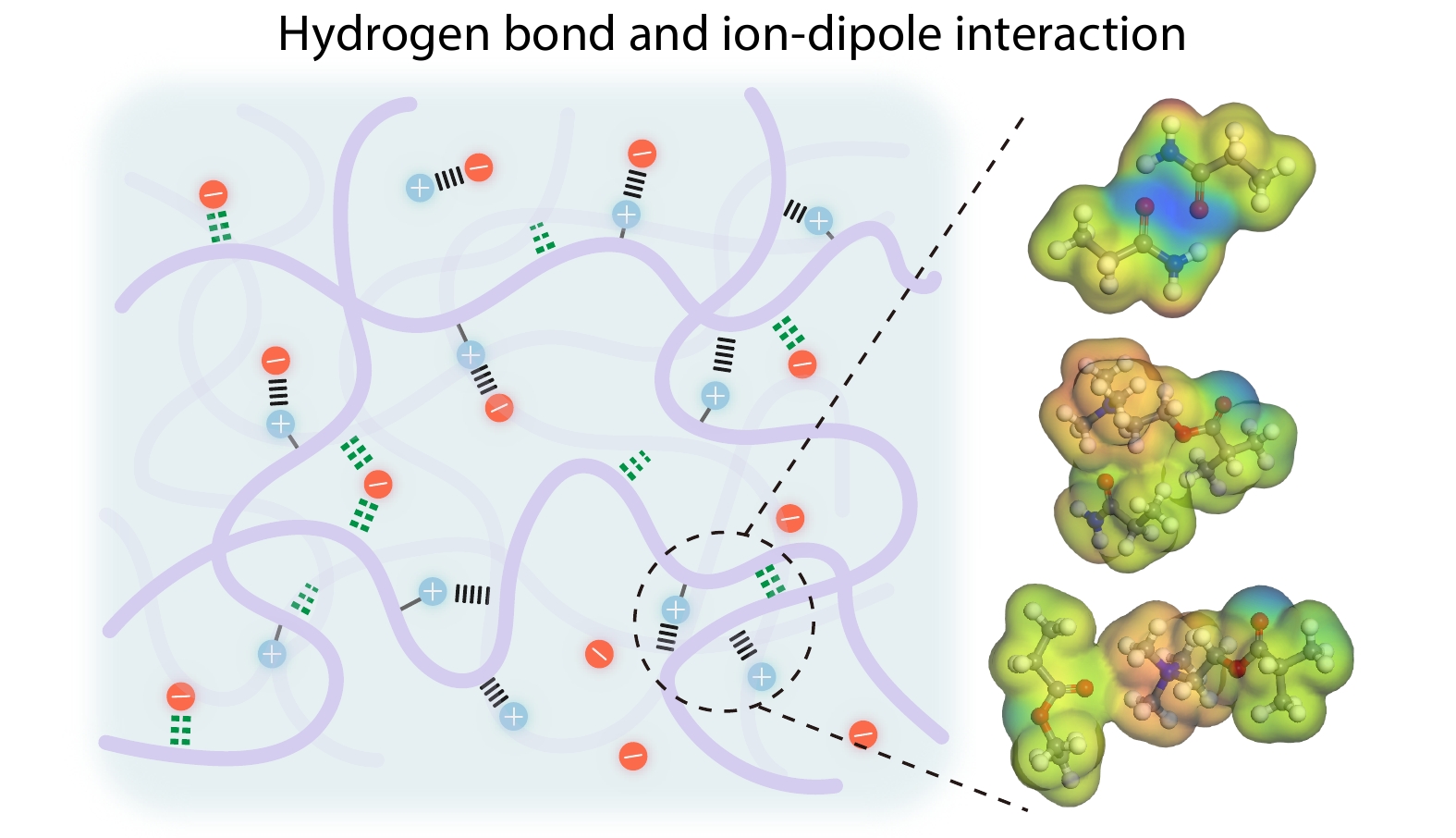
RESEARCH ARTICLE
2025, 43(9): 1496-1504. DOI: 10.1007/s10118-025-3362-2Published(online): 2025-08-25Abstract Full text L-PDFAbstract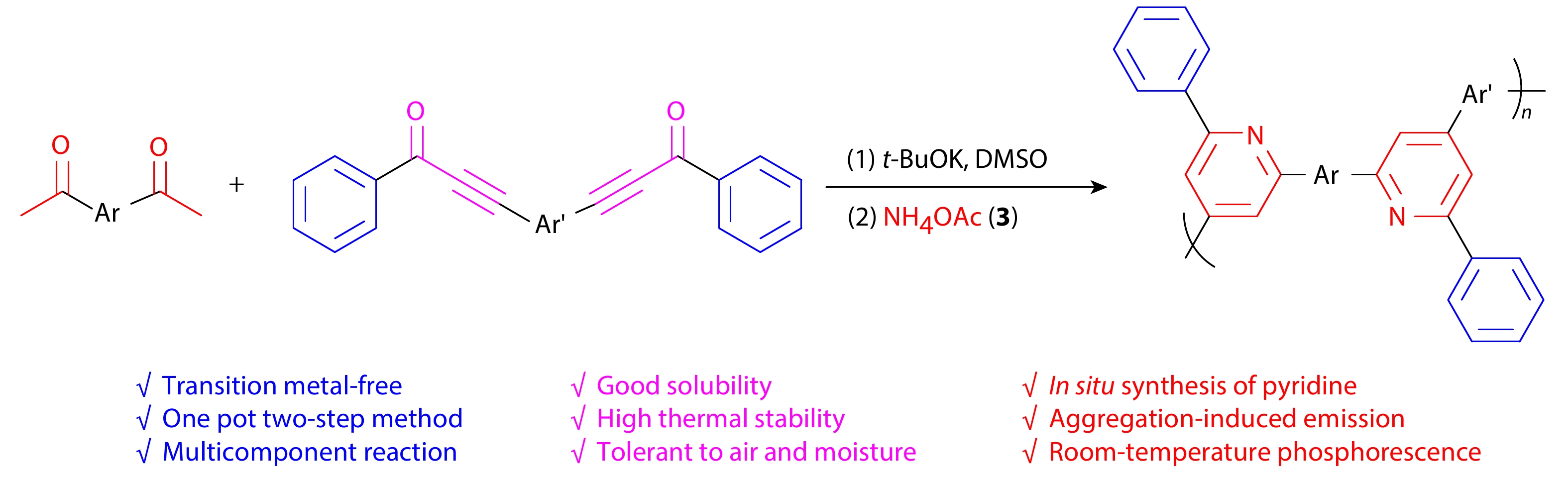
RESEARCH ARTICLE
2025, 43(9): 1505-1515. DOI: 10.1007/s10118-025-3352-4Published(online): 2025-08-25Abstract Full text L-PDFAbstract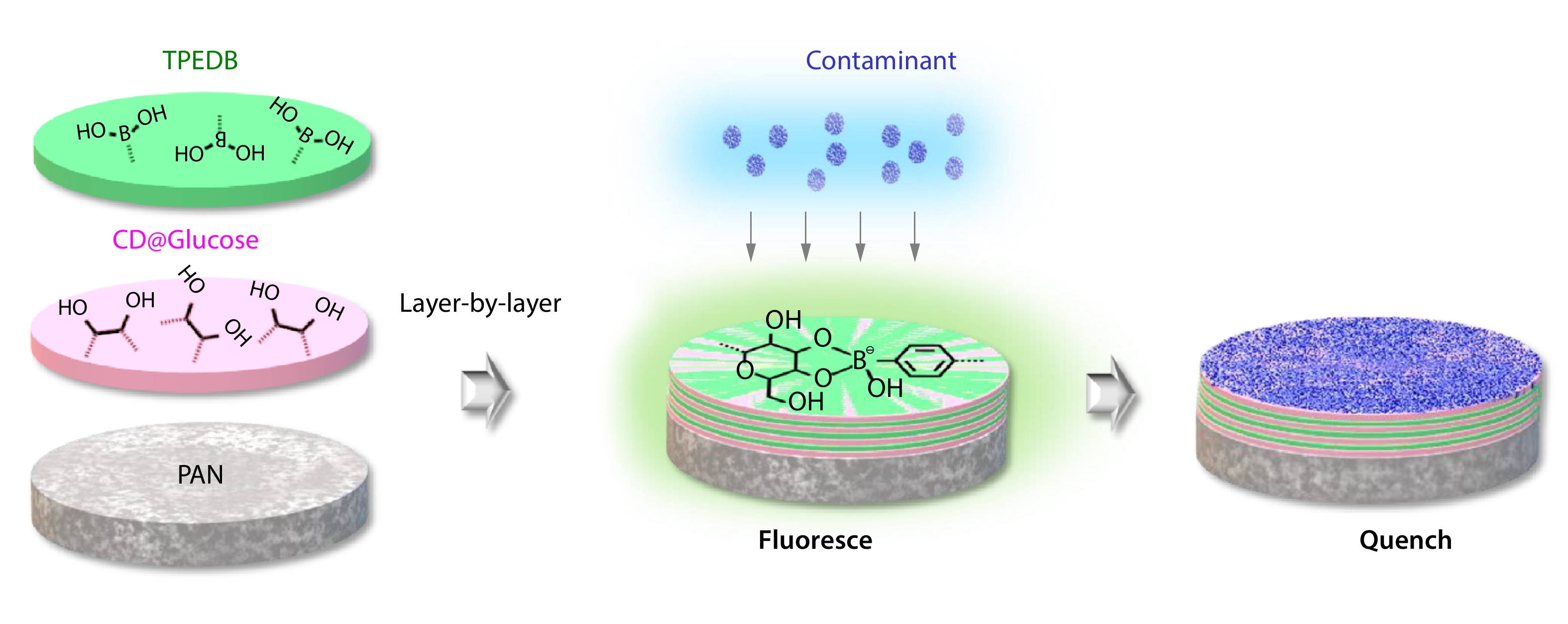
RESEARCH ARTICLE
2025, 43(9): 1516-1526. DOI: 10.1007/s10118-025-3367-xPublished(online): 2025-08-25Abstract Full text L-PDFAbstract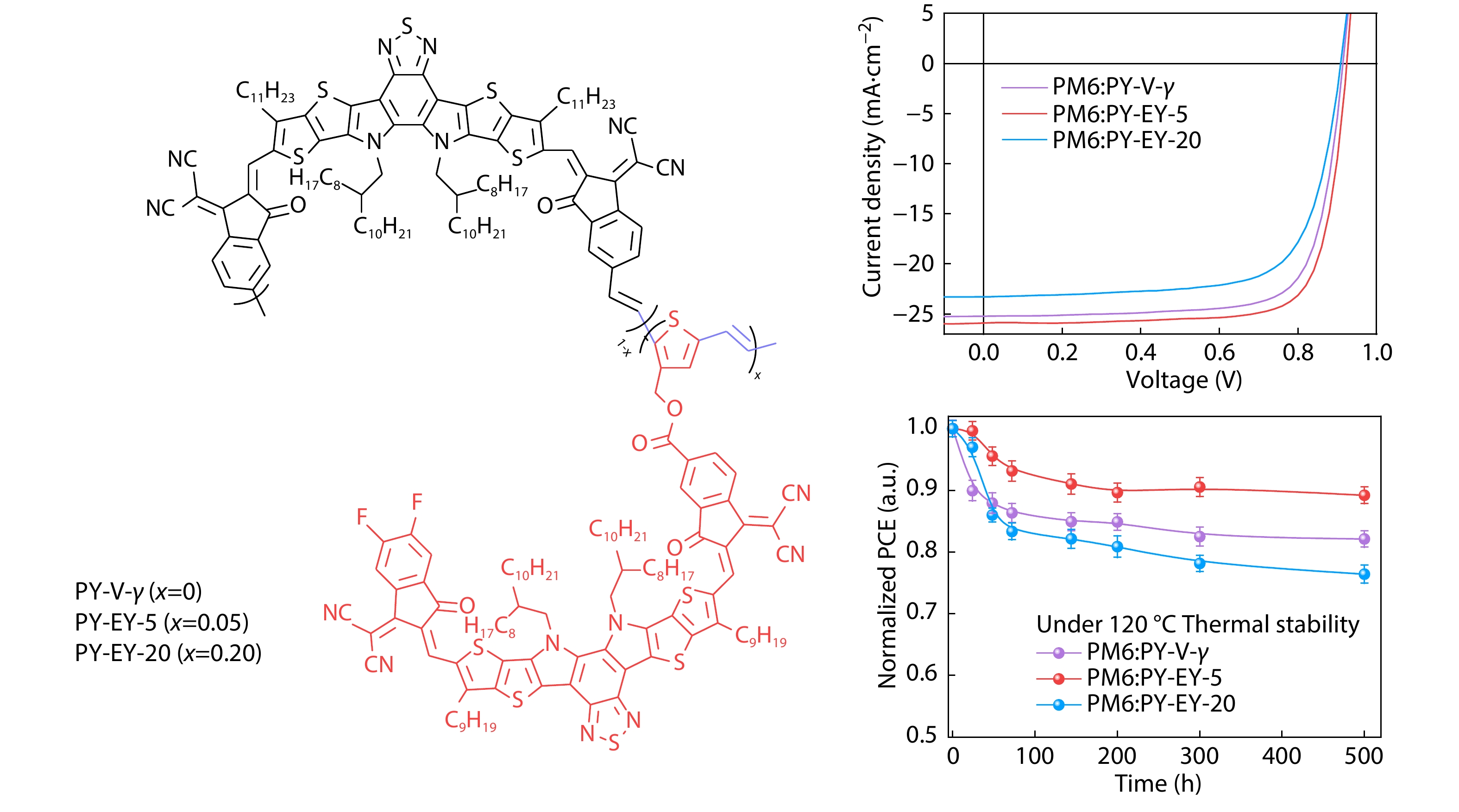
RESEARCH ARTICLE
2025, 43(9): 1527-1536. DOI: 10.1007/s10118-025-3351-5Published(online): 2025-08-25Abstract Full text L-PDFAbstract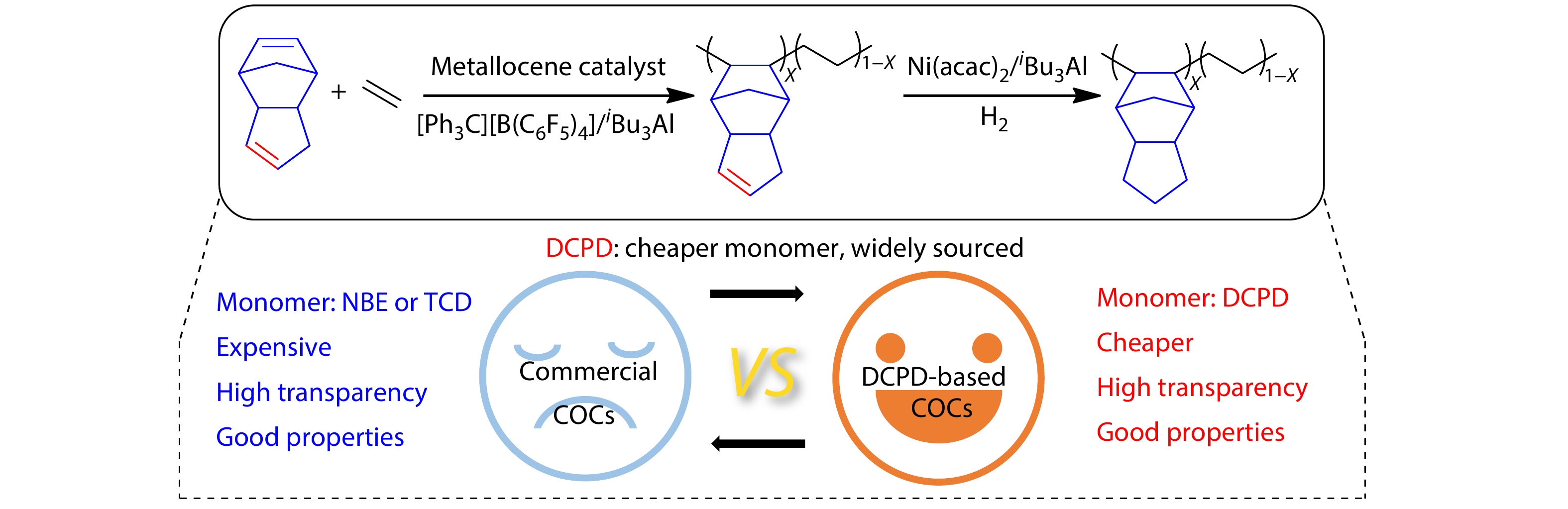
RESEARCH ARTICLE
2025, 43(9): 1537-1548. DOI: 10.1007/s10118-025-3356-0Published(online): 2025-08-25Abstract Full text L-PDFAbstract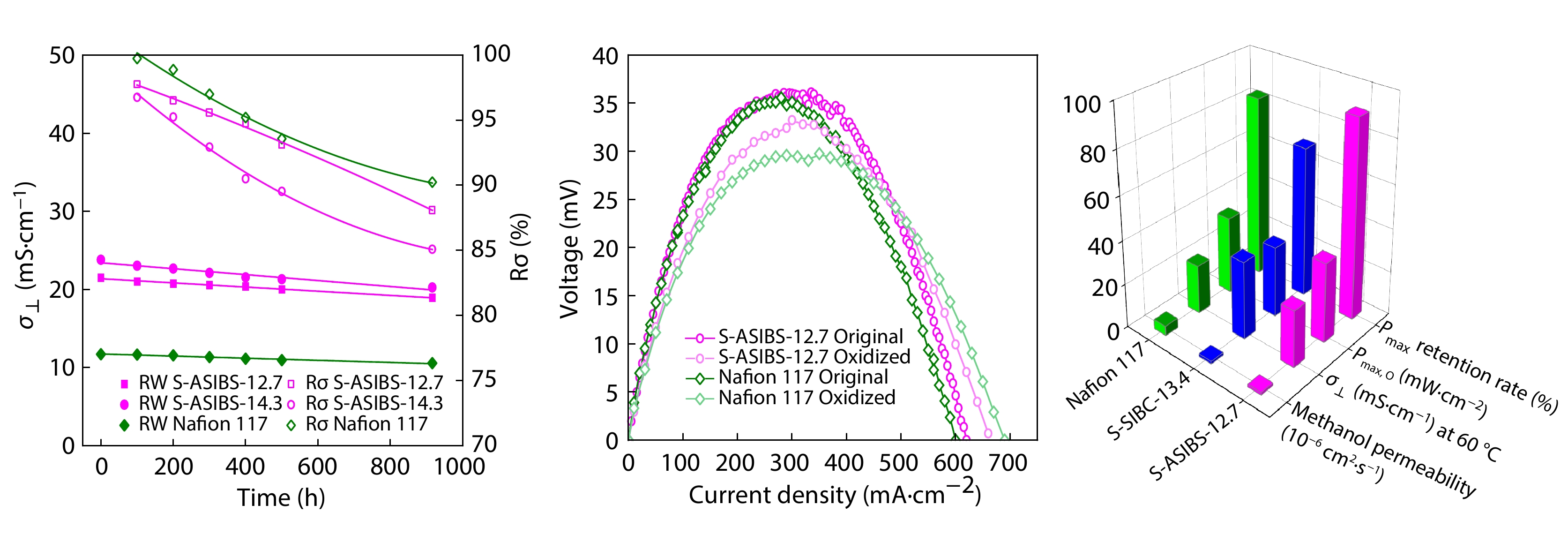
RESEARCH ARTICLE
2025, 43(9): 1549-1564. DOI: 10.1007/s10118-025-3385-8Published(online): 2025-08-25Abstract Full text L-PDFAbstract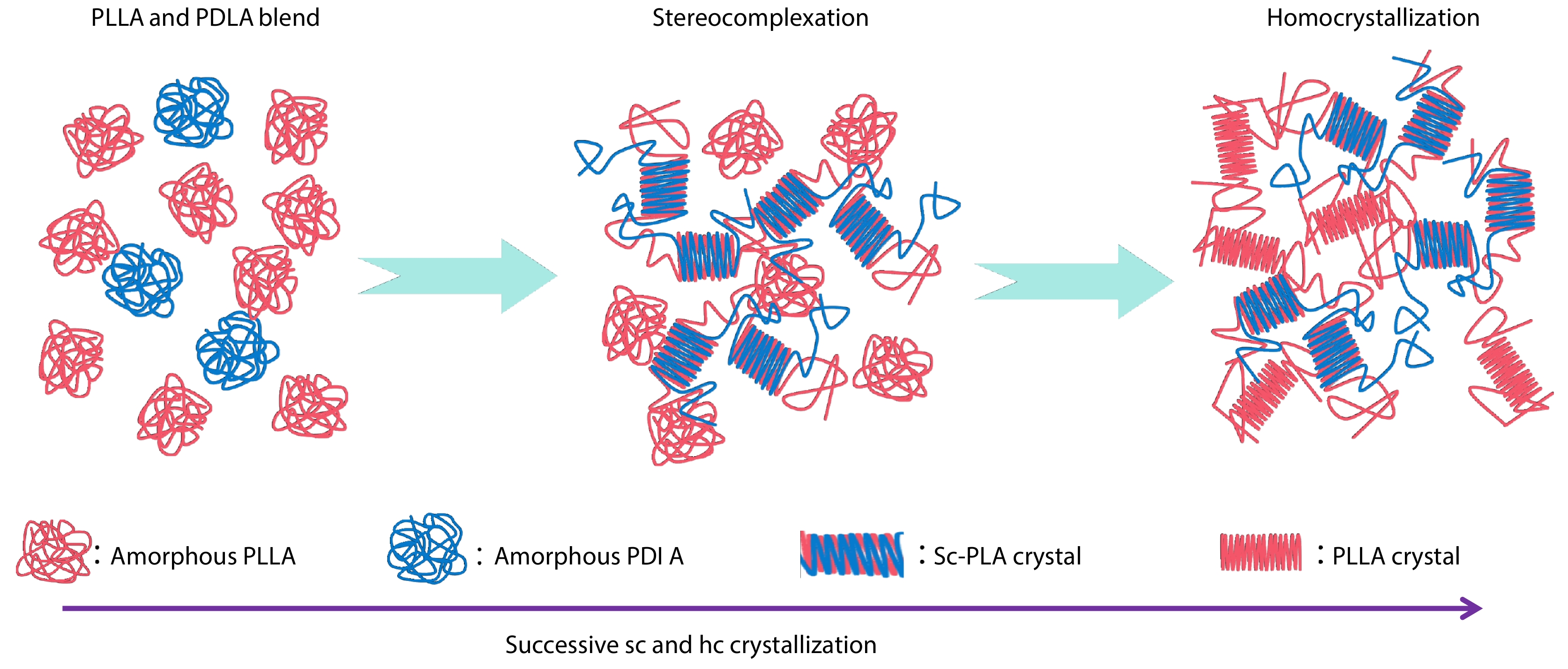
RESEARCH ARTICLE
2025, 43(9): 1565-1575. DOI: 10.1007/s10118-025-3377-8Published(online): 2025-08-25Abstract Full text L-PDFAbstract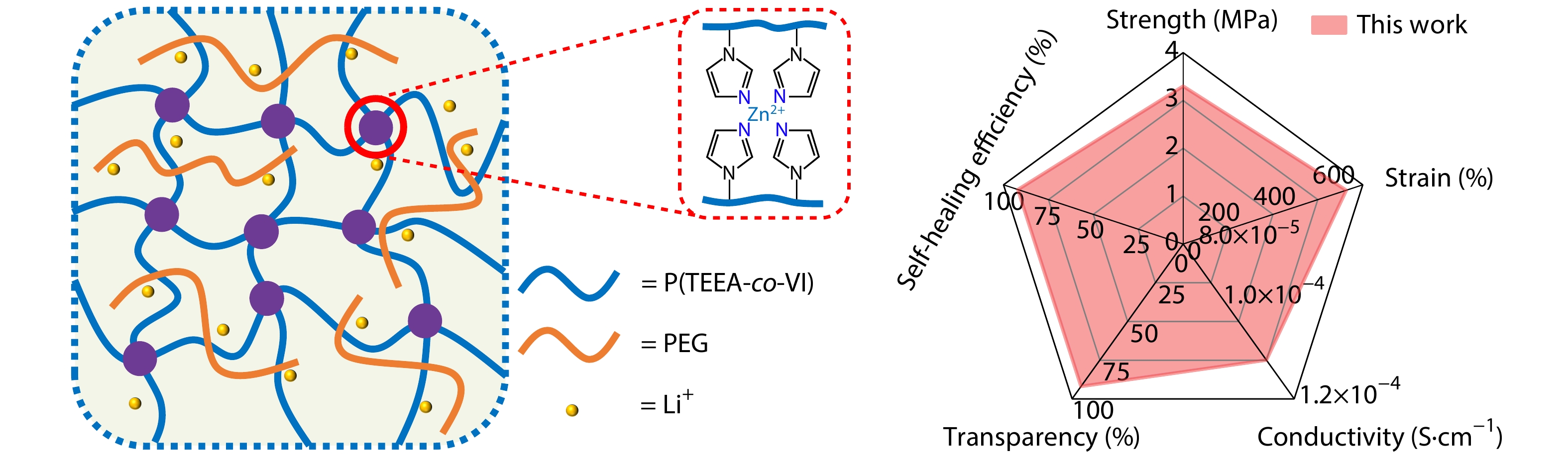
RESEARCH ARTICLE
2025, 43(9): 1576-1583. DOI: 10.1007/s10118-025-3345-3Published(online): 2025-08-25Abstract Full text L-PDFAbstract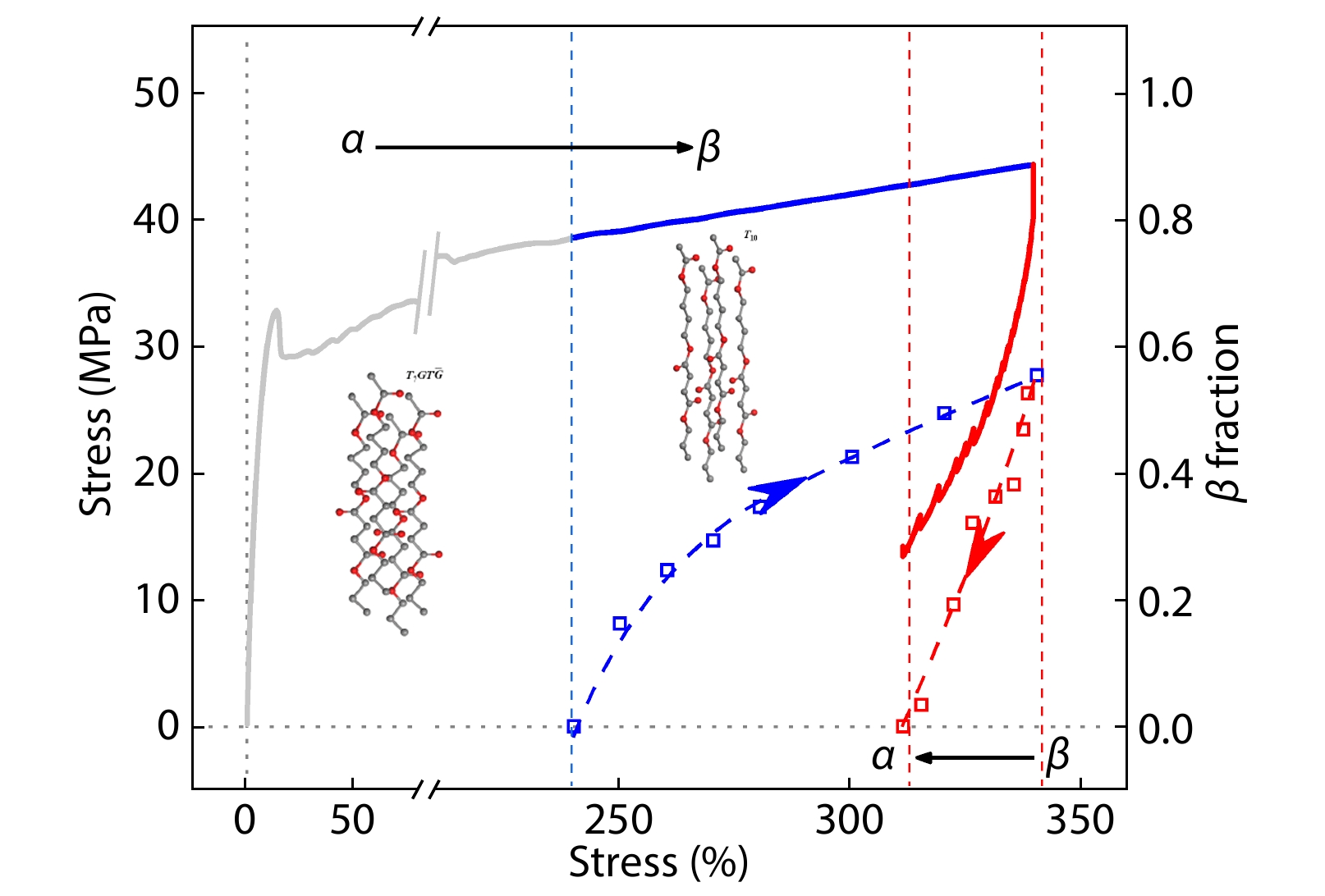
RESEARCH ARTICLE
2025, 43(9): 1584-1591. DOI: 10.1007/s10118-025-3373-zPublished(online): 2025-08-25Abstract Full text L-PDFAbstract
RESEARCH ARTICLE
2025, 43(9): 1592-1601. DOI: 10.1007/s10118-025-3394-7Published(online): 2025-08-25Abstract Full text L-PDFAbstract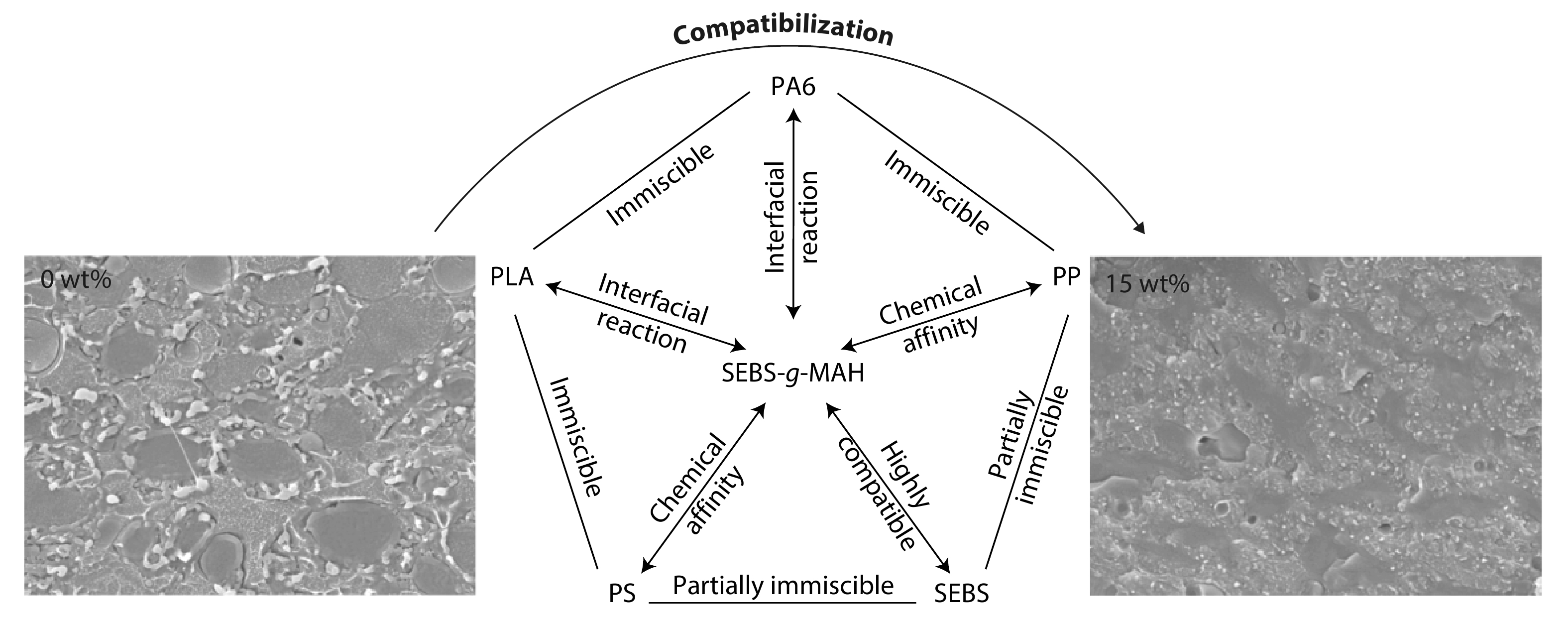
RESEARCH ARTICLE
2025, 43(9): 1602-1615. DOI: 10.1007/s10118-025-3379-6Published(online): 2025-08-25Abstract Full text L-PDFAbstract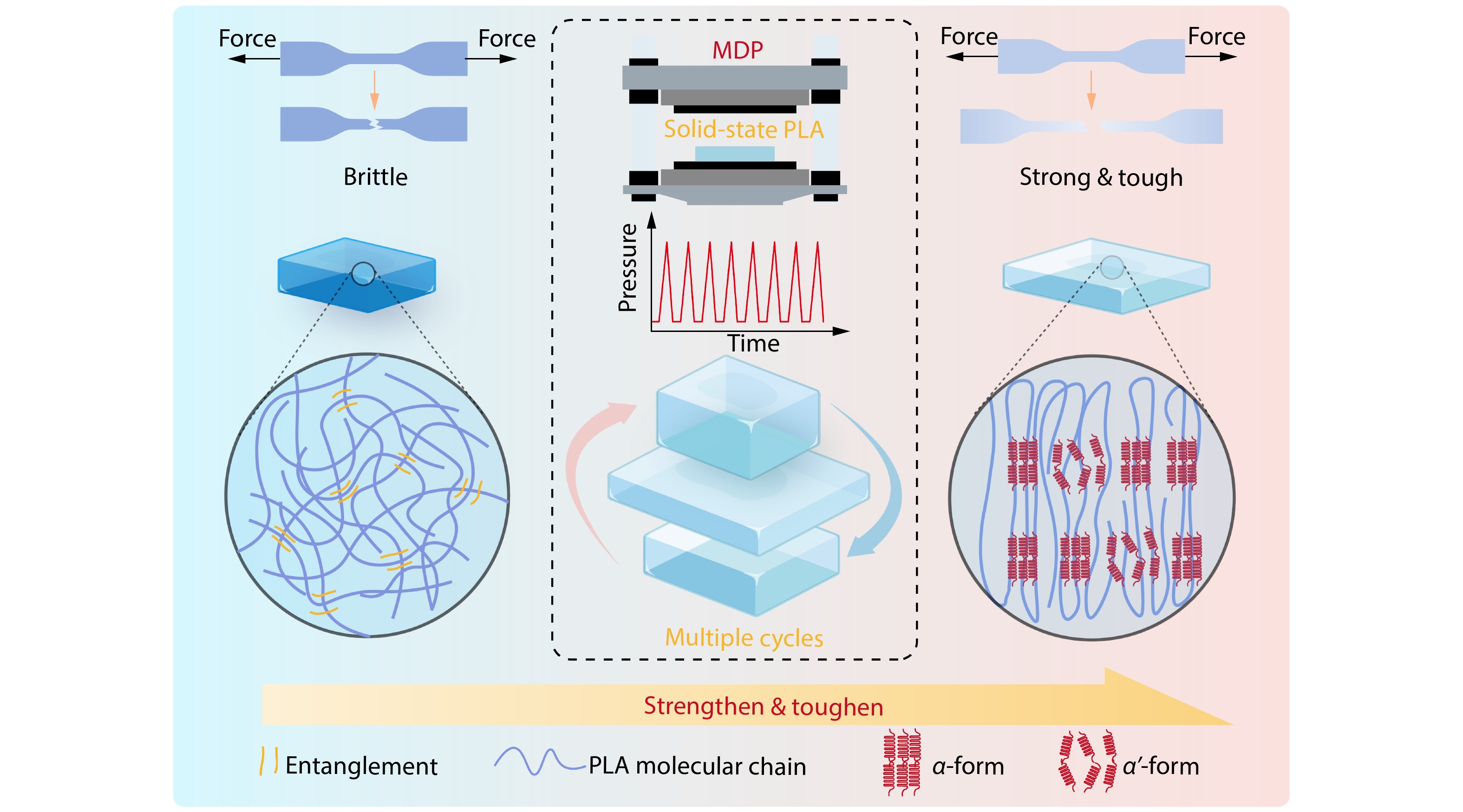
RESEARCH ARTICLE
2025, 43(9): 1616-1628. DOI: 10.1007/s10118-025-3363-1Published(online): 2025-08-25Abstract Full text L-PDFAbstract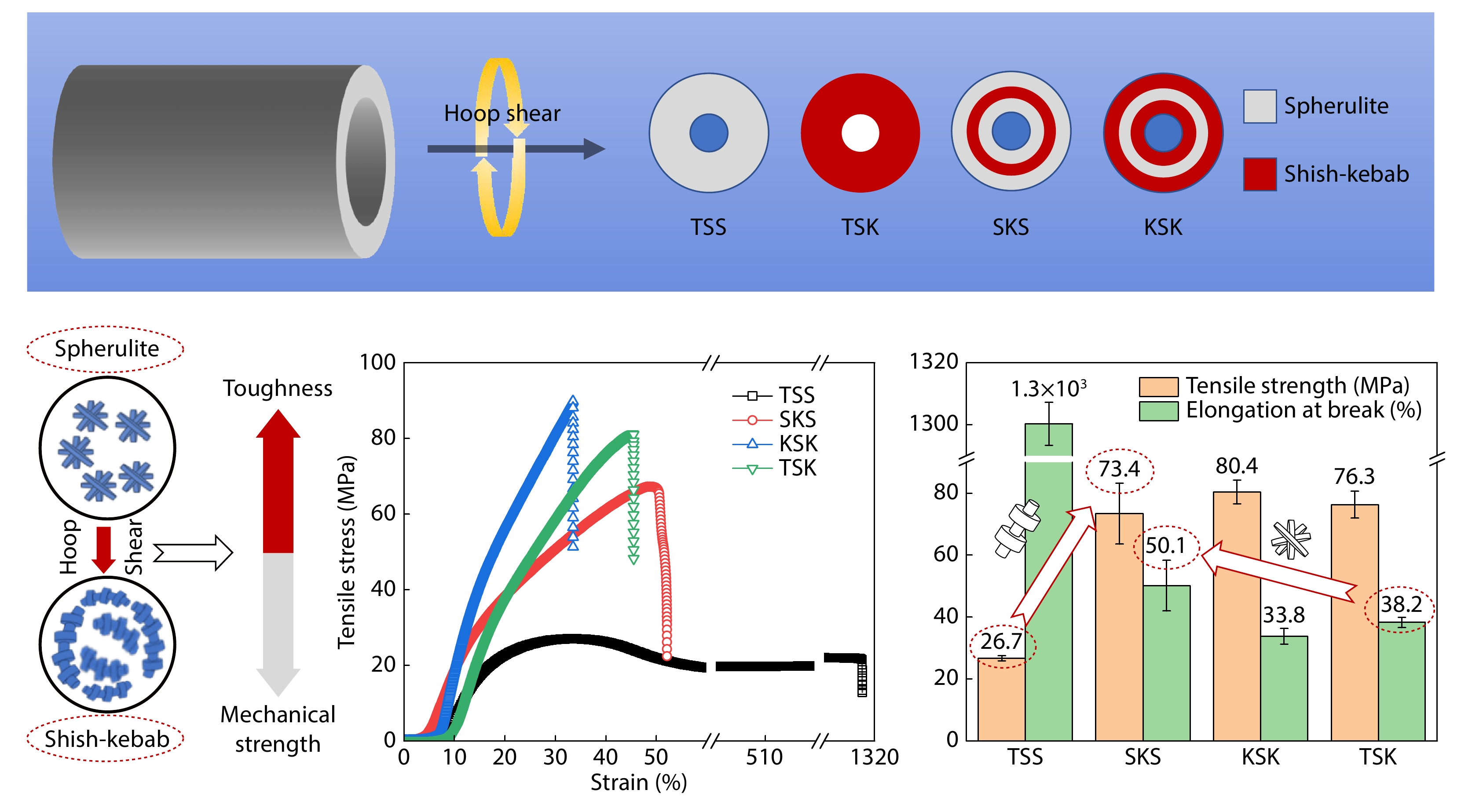
RESEARCH ARTICLE
2025, 43(9): 1629-1637. DOI: 10.1007/s10118-025-3382-yPublished(online): 2025-08-25Abstract Full text L-PDFAbstract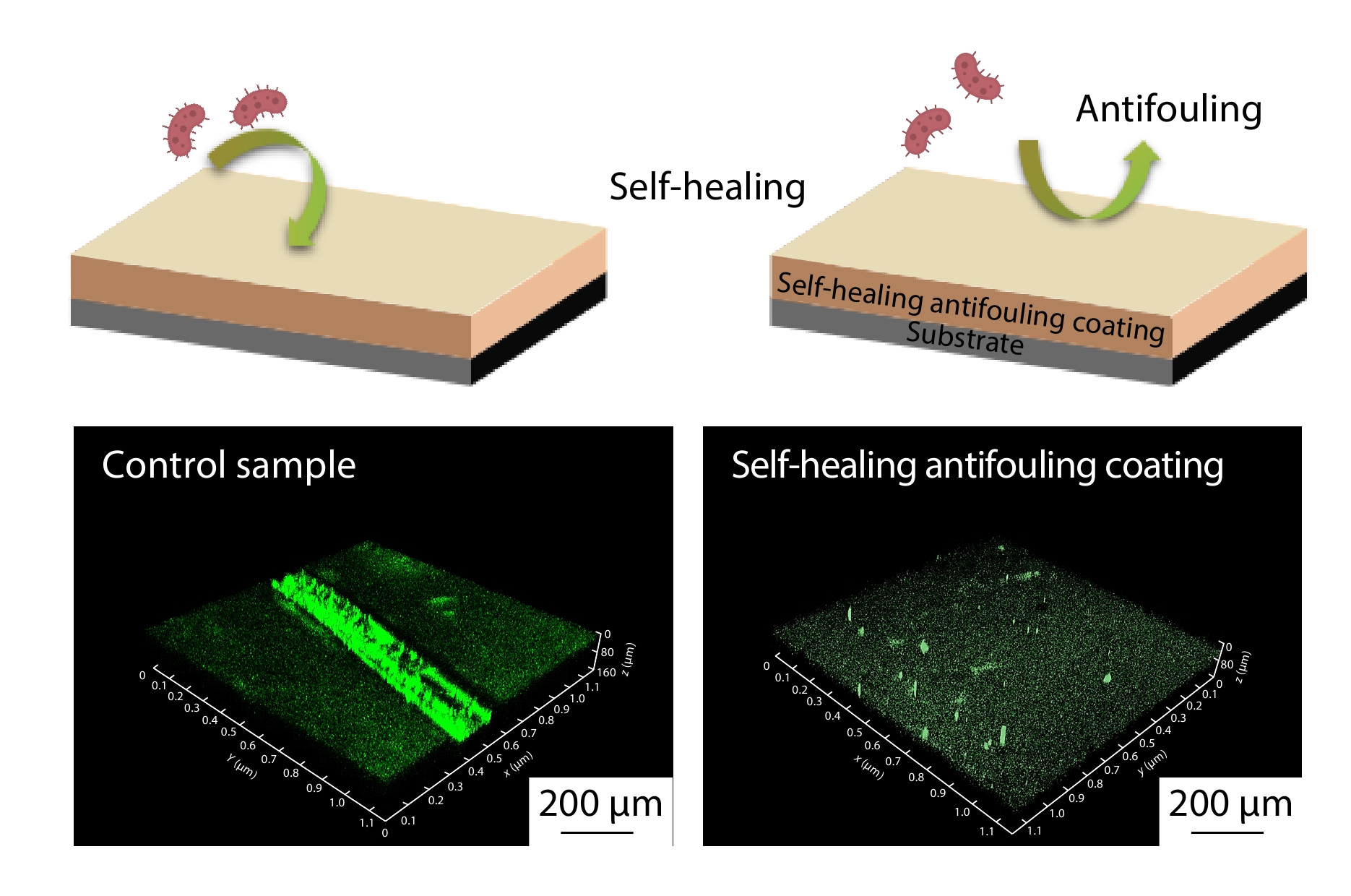
RESEARCH ARTICLE
2025, 43(9): 1638-1650. DOI: 10.1007/s10118-025-3374-yPublished(online): 2025-08-25Abstract Full text L-PDFAbstract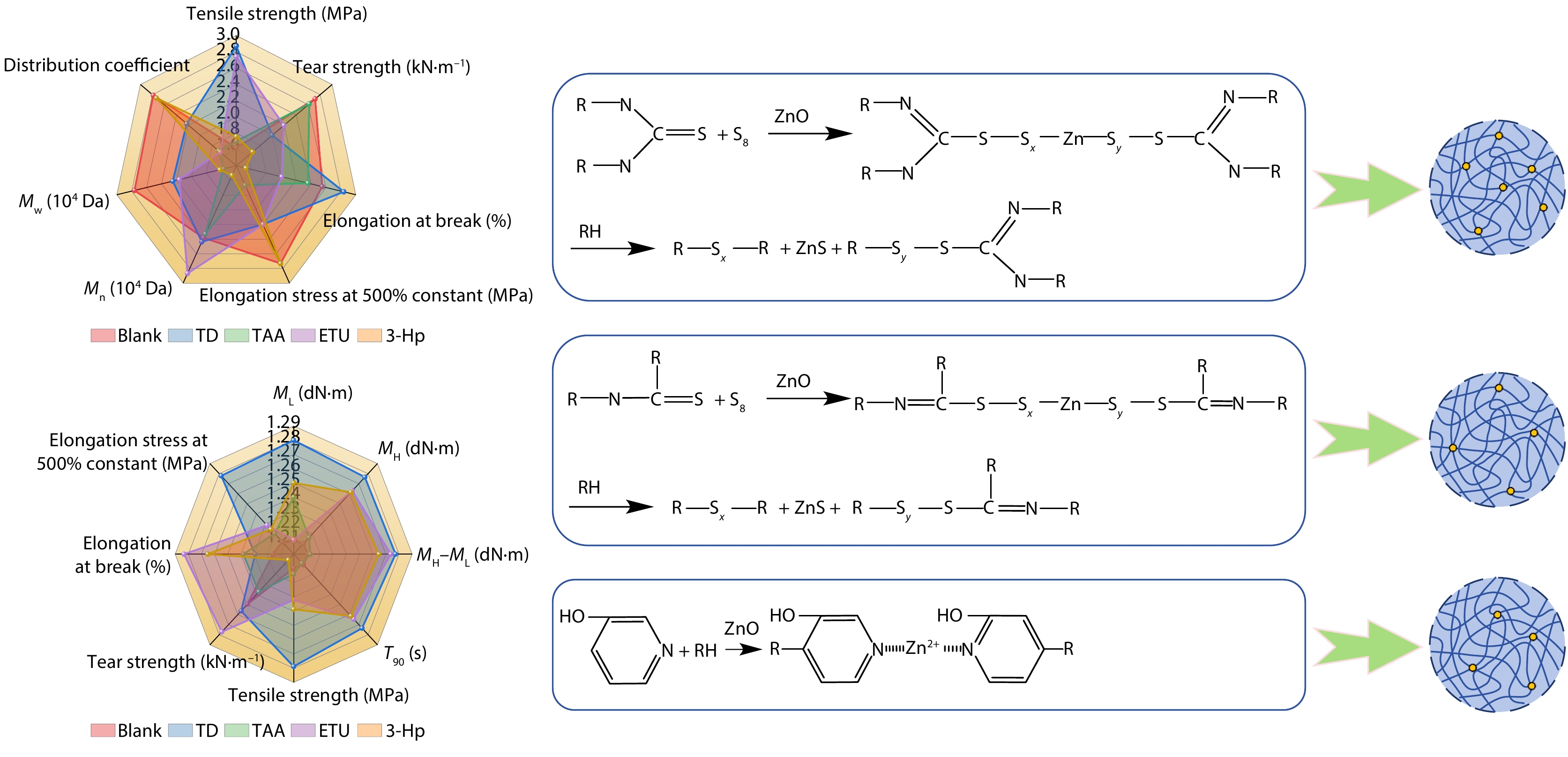
RESEARCH ARTICLE
2025, 43(9): 1651-1660. DOI: 10.1007/s10118-025-3368-9Published(online): 2025-08-25Abstract Full text L-PDFAbstract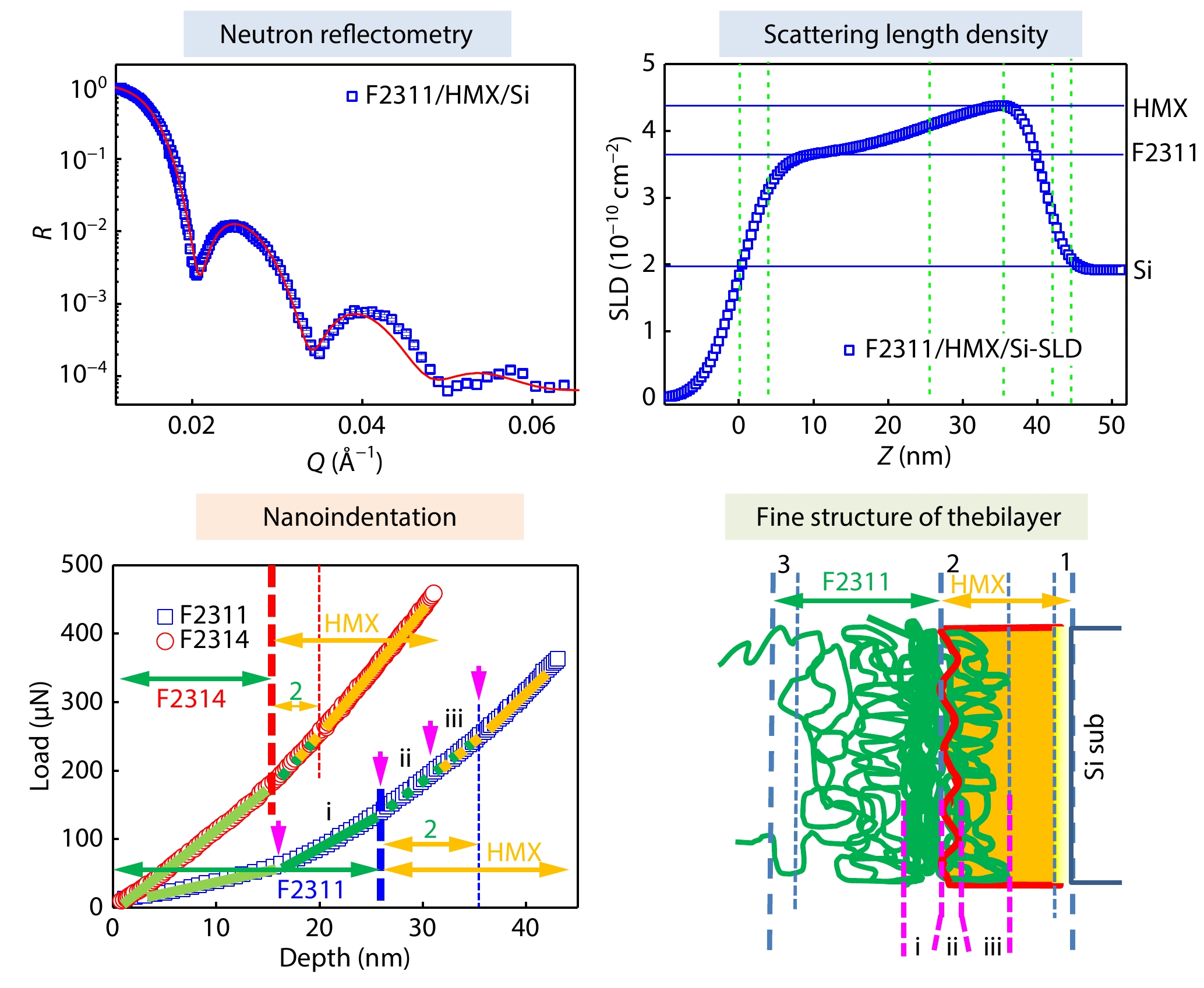
RESEARCH ARTICLE
2025, 43(9): 1661-1670. DOI: 10.1007/s10118-025-3361-3Published(online): 2025-08-25Abstract Full text L-PDFAbstract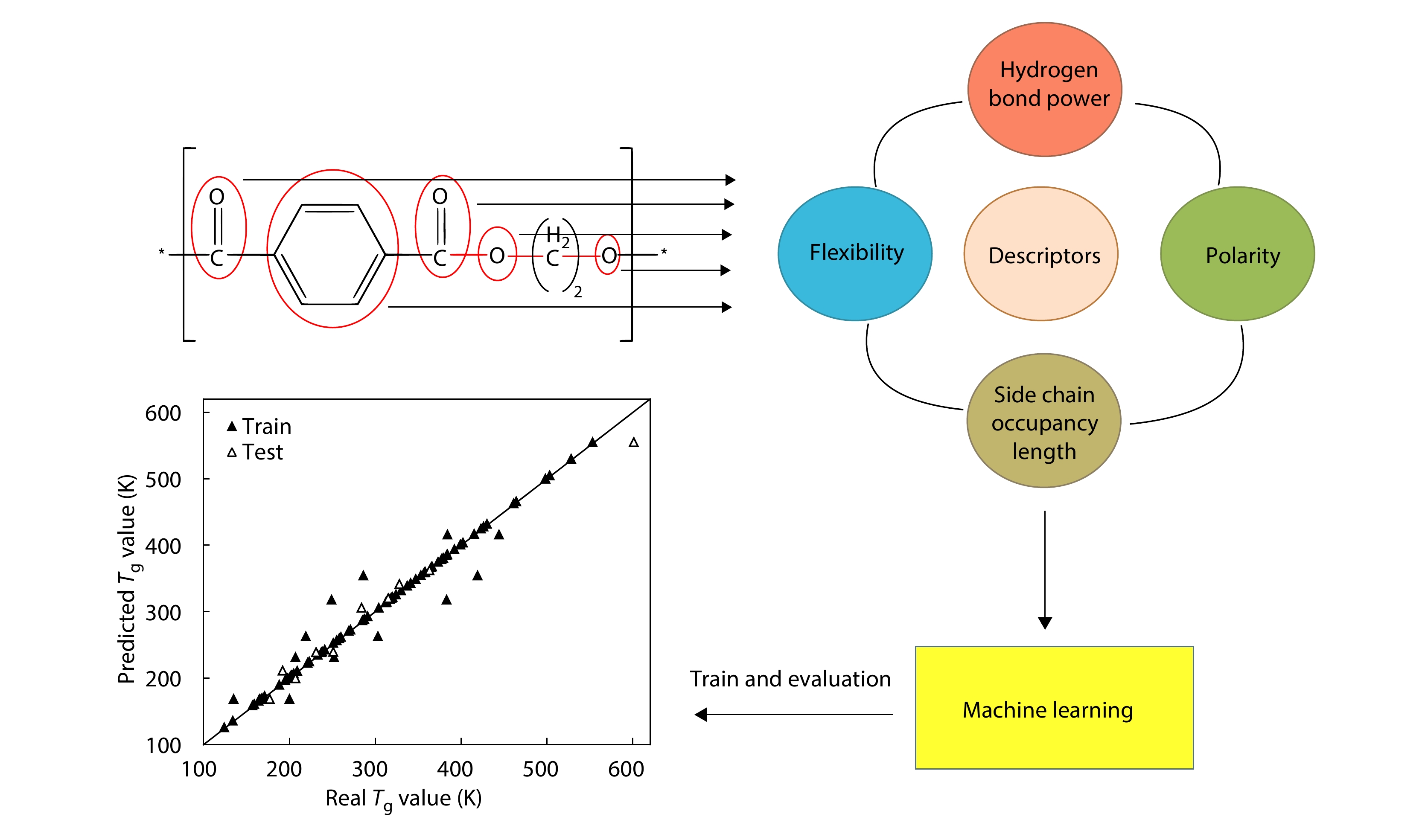
RESEARCH ARTICLE
2025, 43(9): 1671-1680. DOI: 10.1007/s10118-025-3381-zPublished(online): 2025-08-25Abstract Full text L-PDFAbstract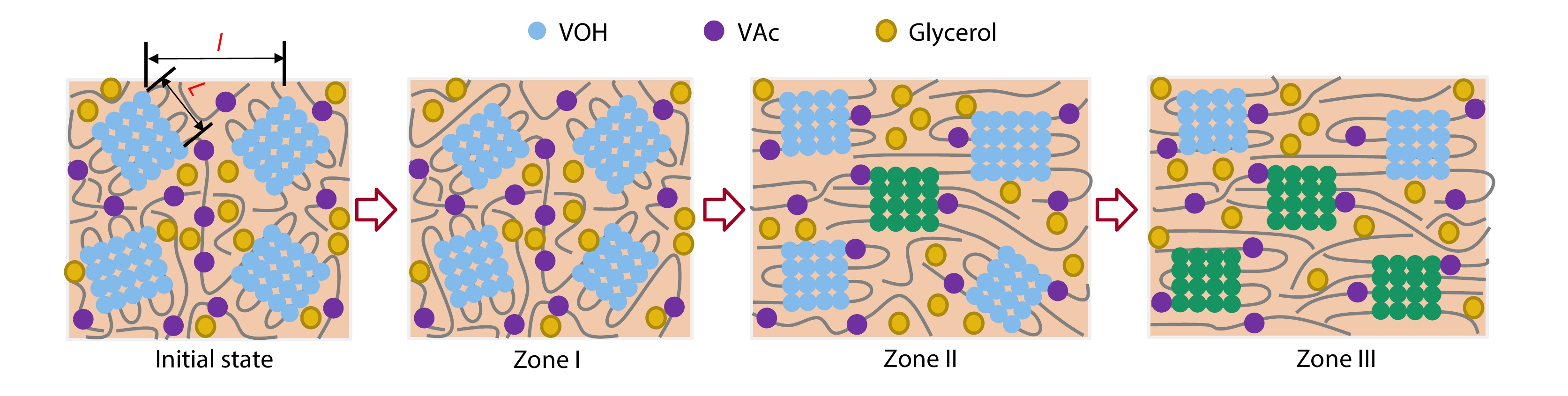
RESEARCH ARTICLE
2025, 43(9): 1681-1689. DOI: 10.1007/s10118-025-3369-8Published(online): 2025-08-25Abstract Full text L-PDFAbstract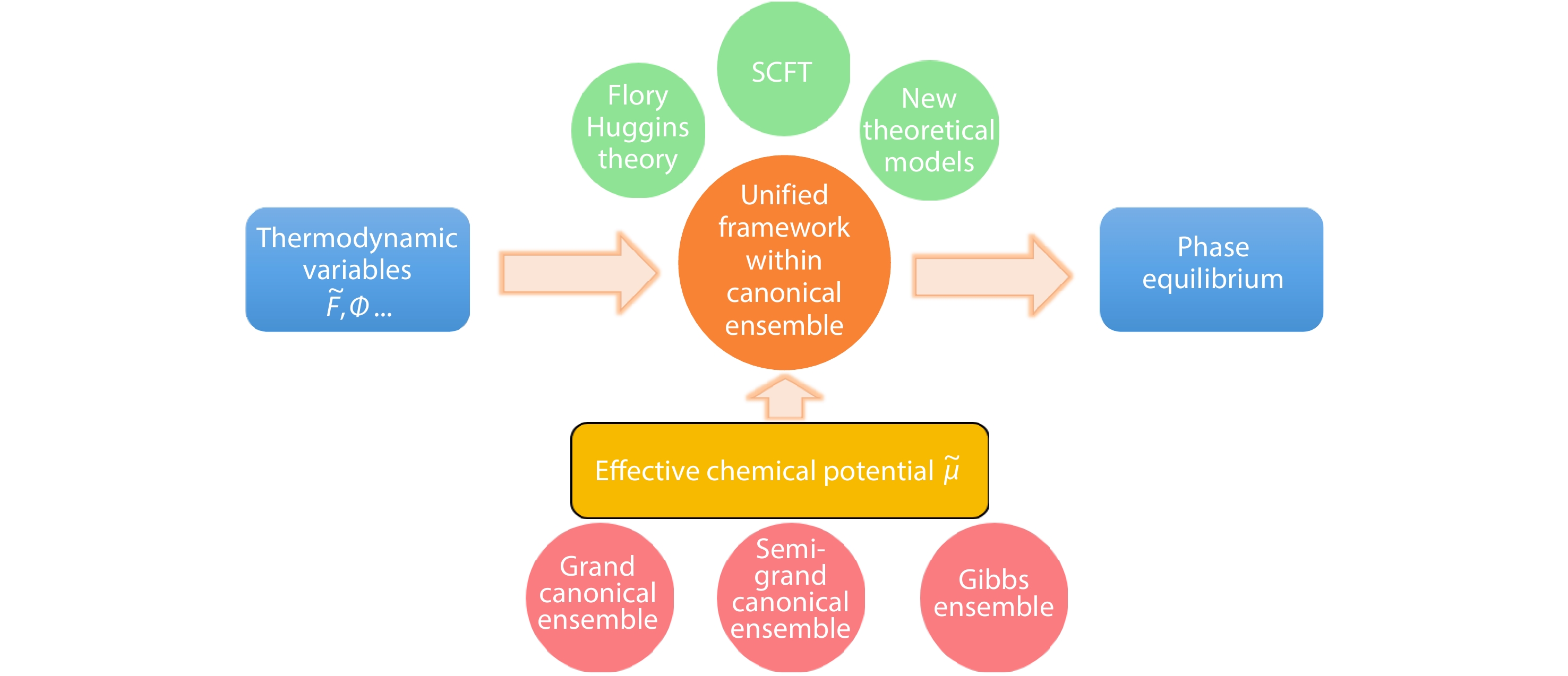
RESEARCH ARTICLE
2025, 43(9): 1690-1698. DOI: 10.1007/s10118-025-3387-6Published(online): 2025-08-25Abstract Full text L-PDFAbstract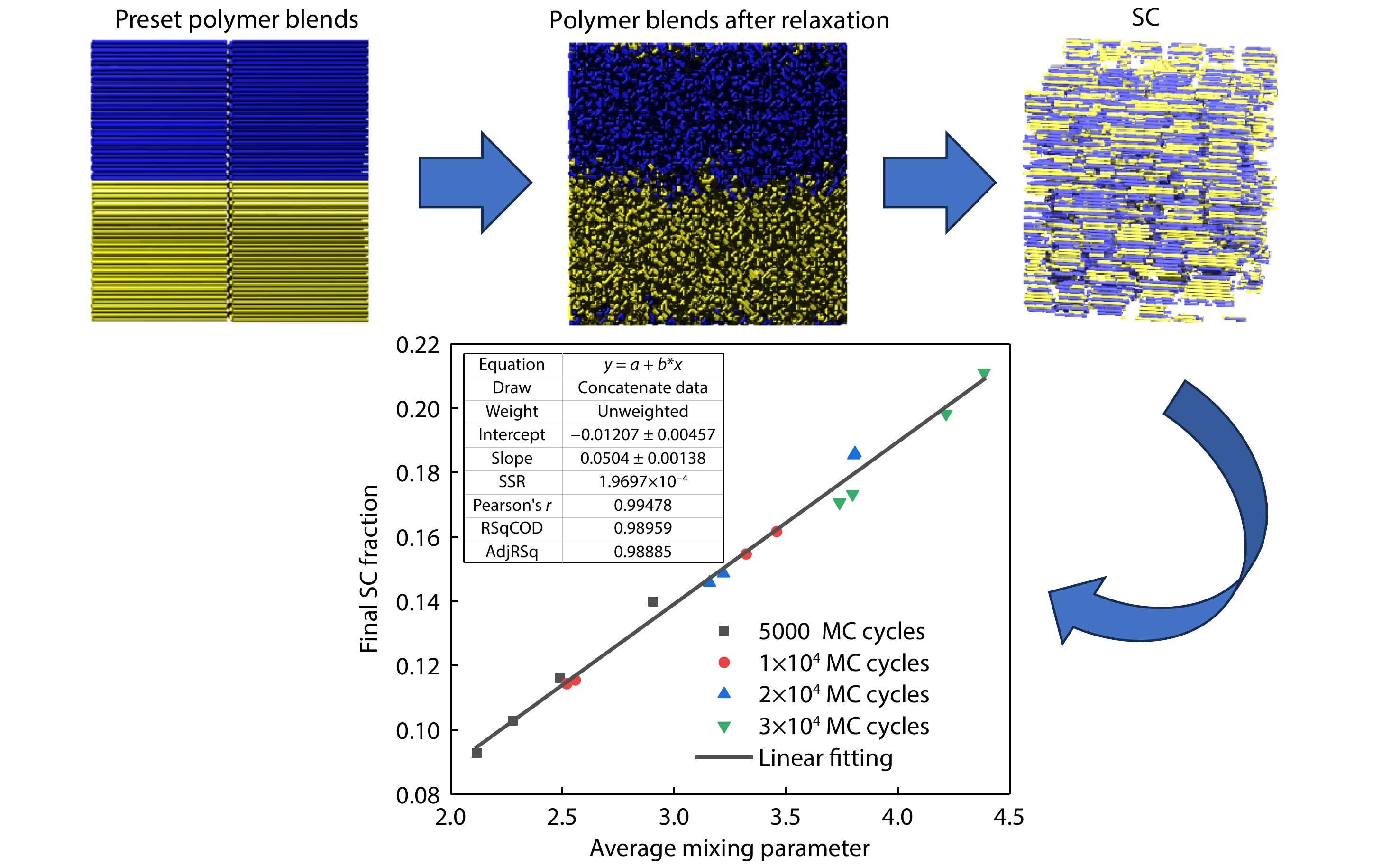

- Technical support is provided by Beijing Founder electronics co., LTD 京ICP备09064830号-19
 京公网安备11010802024621
京公网安备11010802024621 - It is recommended to read the content of this site in Chrome&IE9+. Please switch to extreme mode in browser 360.
- Cookies We use cookies to help provide and enhance our service and tailor content. By continuing, you agree to the use of cookies.
0