
Latest Issue
Just Accepted Online First List of Issues Document Types
Issue 6, 2025
PREFACE
2025, 43(6): 875. DOI: 10.1007/s10118-025-3383-xPublished(online): 2025-07-14Full text L-PDF

REVIEW
2025, 43(6): 876-886. DOI: 10.1007/s10118-024-3226-1Published(online): 2025-07-14Abstract Full text L-PDFAbstract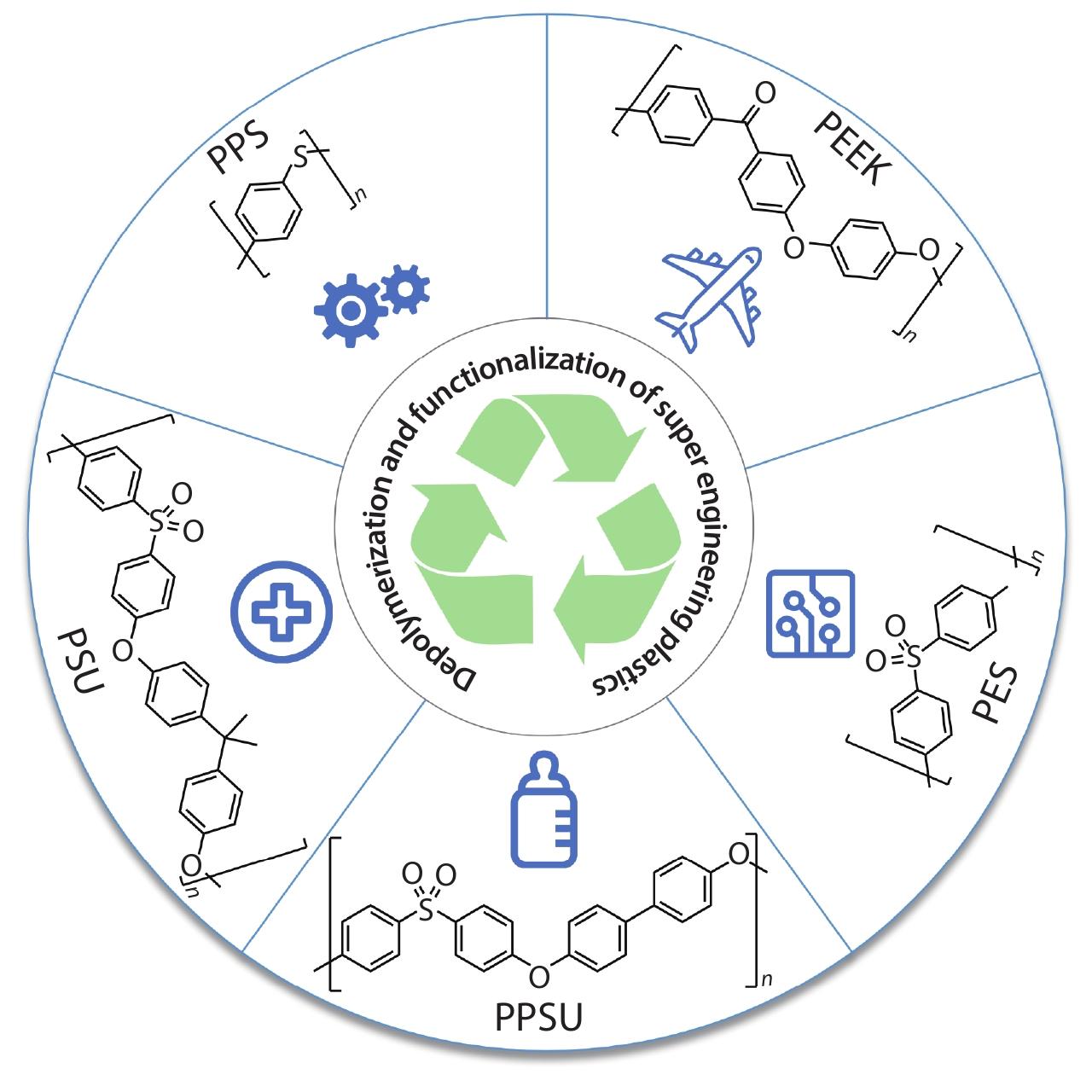
REVIEW
2025, 43(6): 887-907. DOI: 10.1007/s10118-025-3278-xPublished(online): 2025-07-14Abstract Full text L-PDFAbstract
RESEARCH ARTICLE
2025, 43(6): 908-913. DOI: 10.1007/s10118-024-3220-7Published(online): 2025-07-14Abstract Full text L-PDFAbstract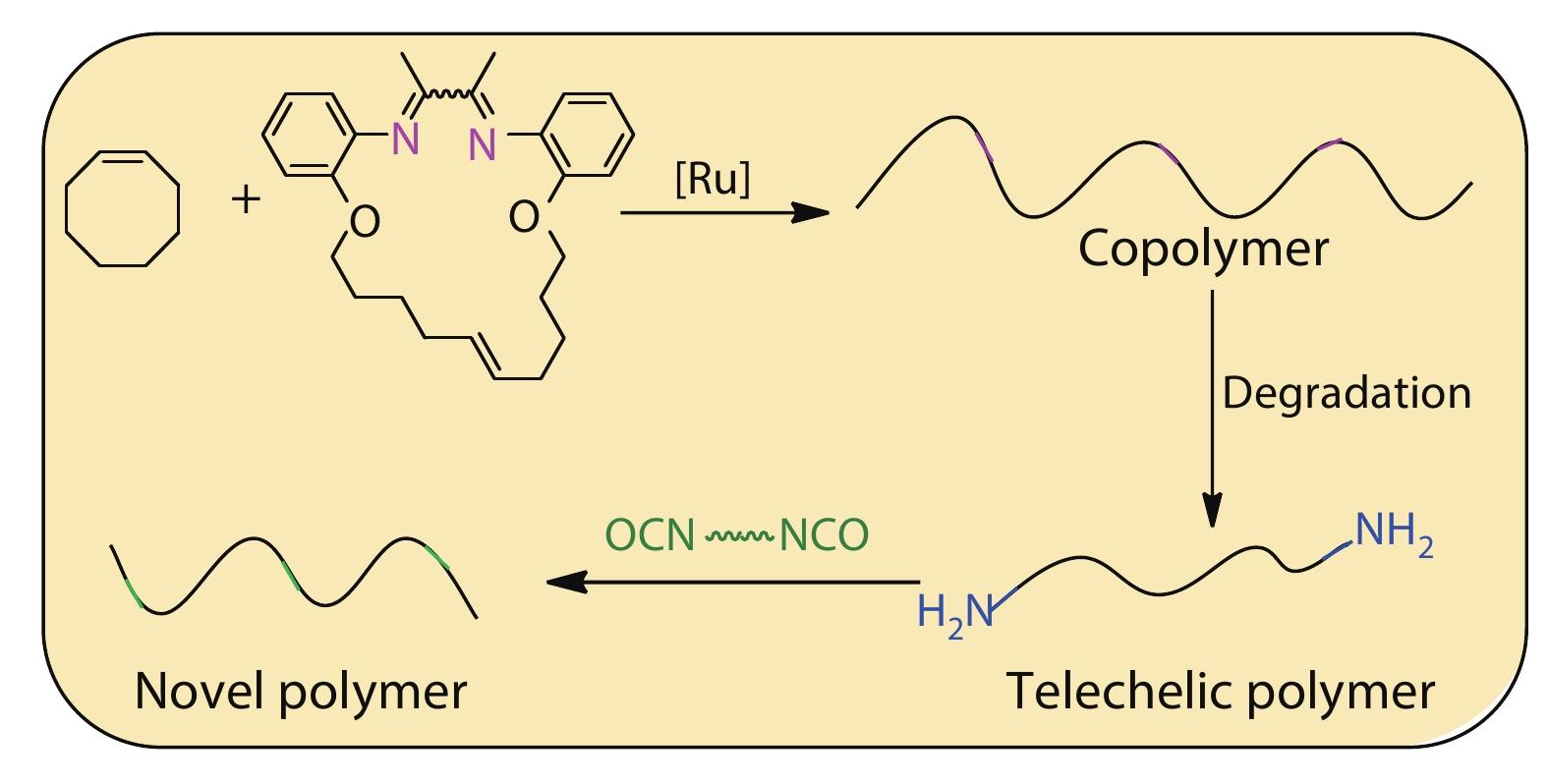
RESEARCH ARTICLE
2025, 43(6): 914-923. DOI: 10.1007/s10118-025-3263-4Published(online): 2025-07-14Abstract Full text L-PDFAbstract
RESEARCH ARTICLE
2025, 43(6): 924-932. DOI: 10.1007/s10118-025-3262-5Published(online): 2025-07-14Abstract Full text L-PDFAbstract
RESEARCH ARTICLE
2025, 43(6): 933-945. DOI: 10.1007/s10118-025-3267-0Published(online): 2025-07-14Abstract Full text L-PDFAbstract
RESEARCH ARTICLE
2025, 43(6): 946-951. DOI: 10.1007/s10118-025-3266-1Published(online): 2025-07-14Abstract Full text L-PDFAbstract
RESEARCH ARTICLE
2025, 43(6): 952-957. DOI: 10.1007/s10118-025-3285-yPublished(online): 2025-07-14Abstract Full text L-PDFAbstract
RESEARCH ARTICLE
2025, 43(6): 958-963. DOI: 10.1007/s10118-025-3280-3Published(online): 2025-07-14Abstract Full text L-PDFAbstract
RESEARCH ARTICLE
2025, 43(6): 964-972. DOI: 10.1007/s10118-025-3297-7Published(online): 2025-07-14Abstract Full text L-PDFAbstract
RESEARCH ARTICLE
2025, 43(6): 973-980. DOI: 10.1007/s10118-025-3372-0Published(online): 2025-07-14Abstract Full text L-PDFAbstract
RESEARCH ARTICLE
2025, 43(6): 981-993. DOI: 10.1007/s10118-025-3378-7Published(online): 2025-07-14Abstract Full text L-PDFAbstract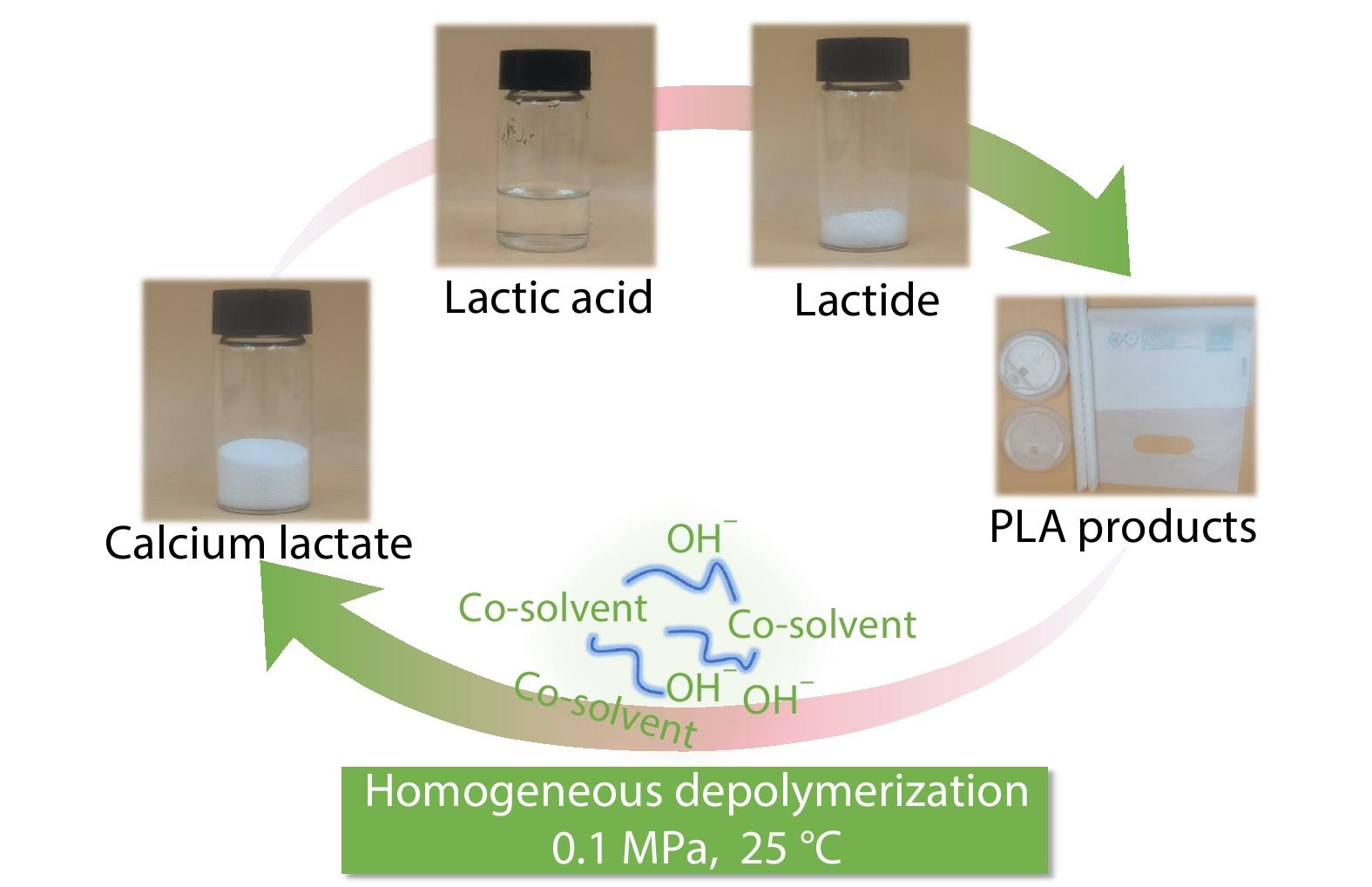
RESEARCH ARTICLE
2025, 43(6): 994-1000. DOI: 10.1007/s10118-025-3342-6Published(online): 2025-07-14Abstract Full text L-PDFAbstract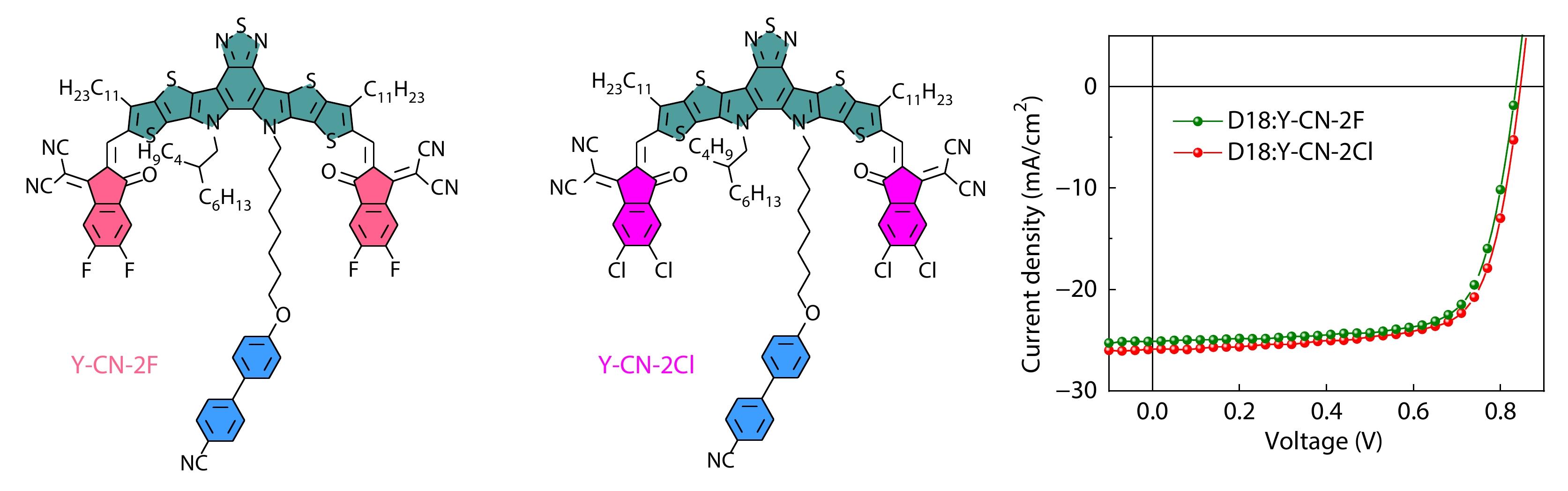
RESEARCH ARTICLE
2025, 43(6): 1001-1011. DOI: 10.1007/s10118-025-3315-9Published(online): 2025-07-14Abstract Full text L-PDFAbstract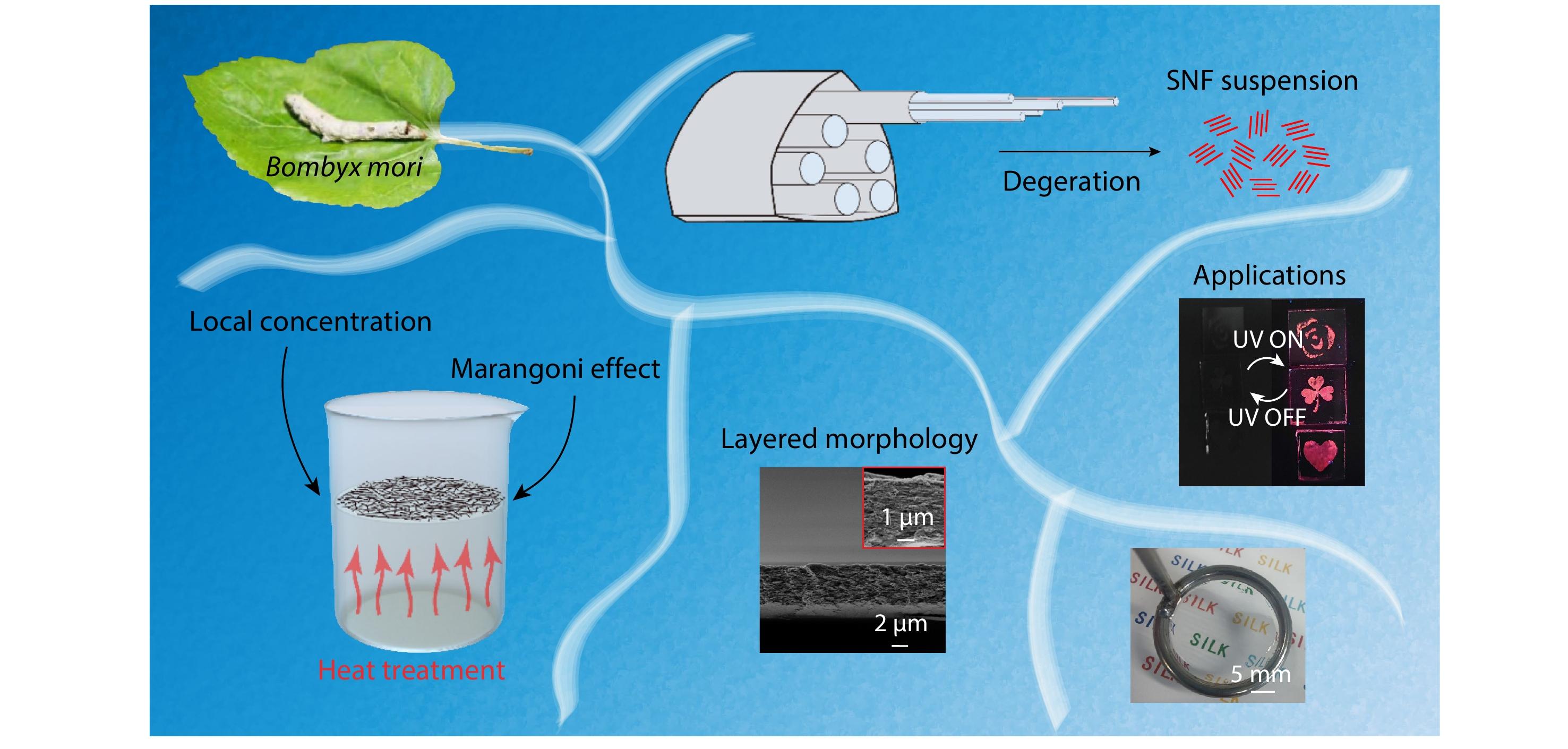
RESEARCH ARTICLE
2025, 43(6): 1012-1021. DOI: 10.1007/s10118-025-3322-xPublished(online): 2025-07-14Abstract Full text L-PDFAbstract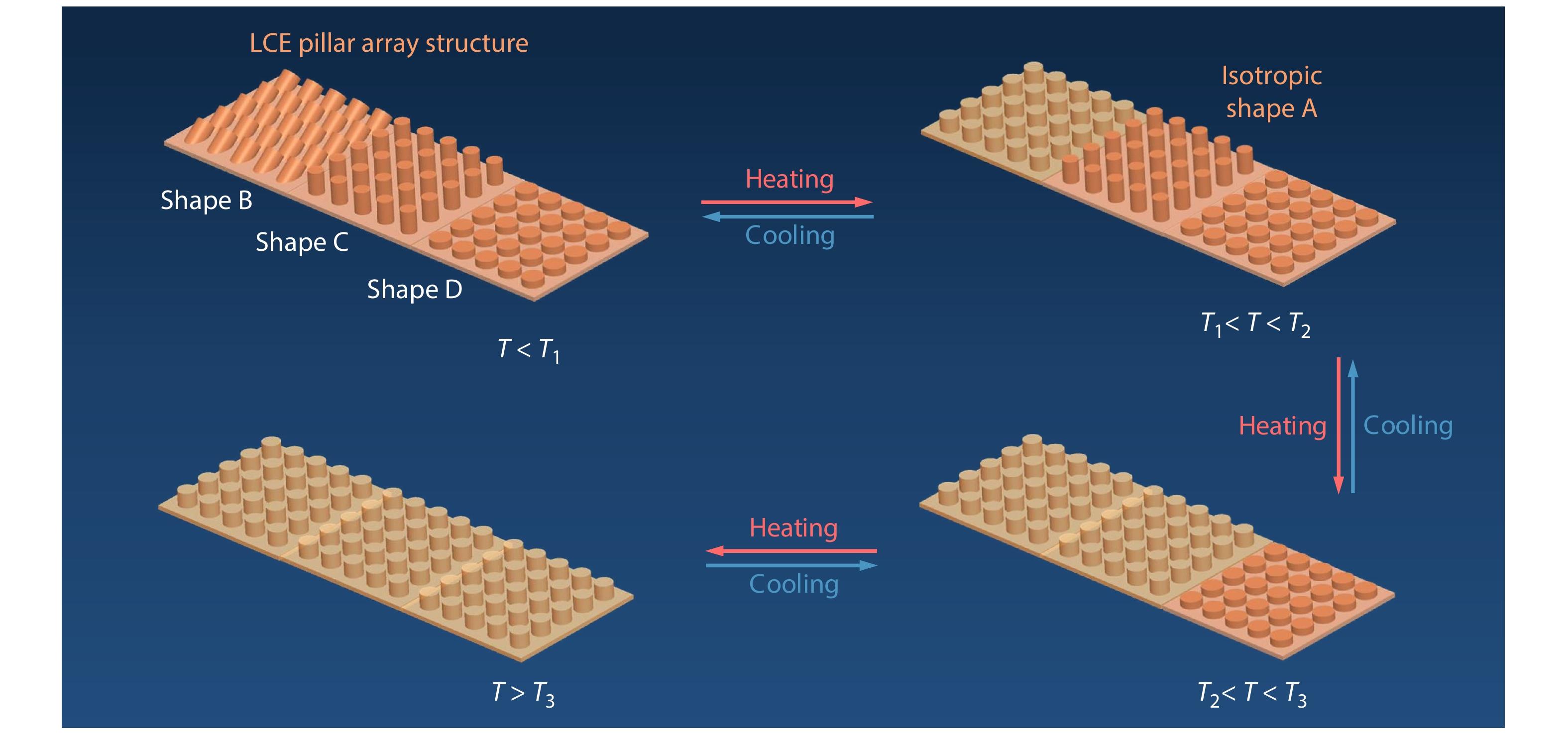
RESEARCH ARTICLE
2025, 43(6): 1022-1031. DOI: 10.1007/s10118-025-3333-7Published(online): 2025-07-14Abstract Full text L-PDFAbstract
RESEARCH ARTICLE
2025, 43(6): 1032-1042. DOI: 10.1007/s10118-025-3325-7Published(online): 2025-07-14Abstract Full text L-PDFAbstract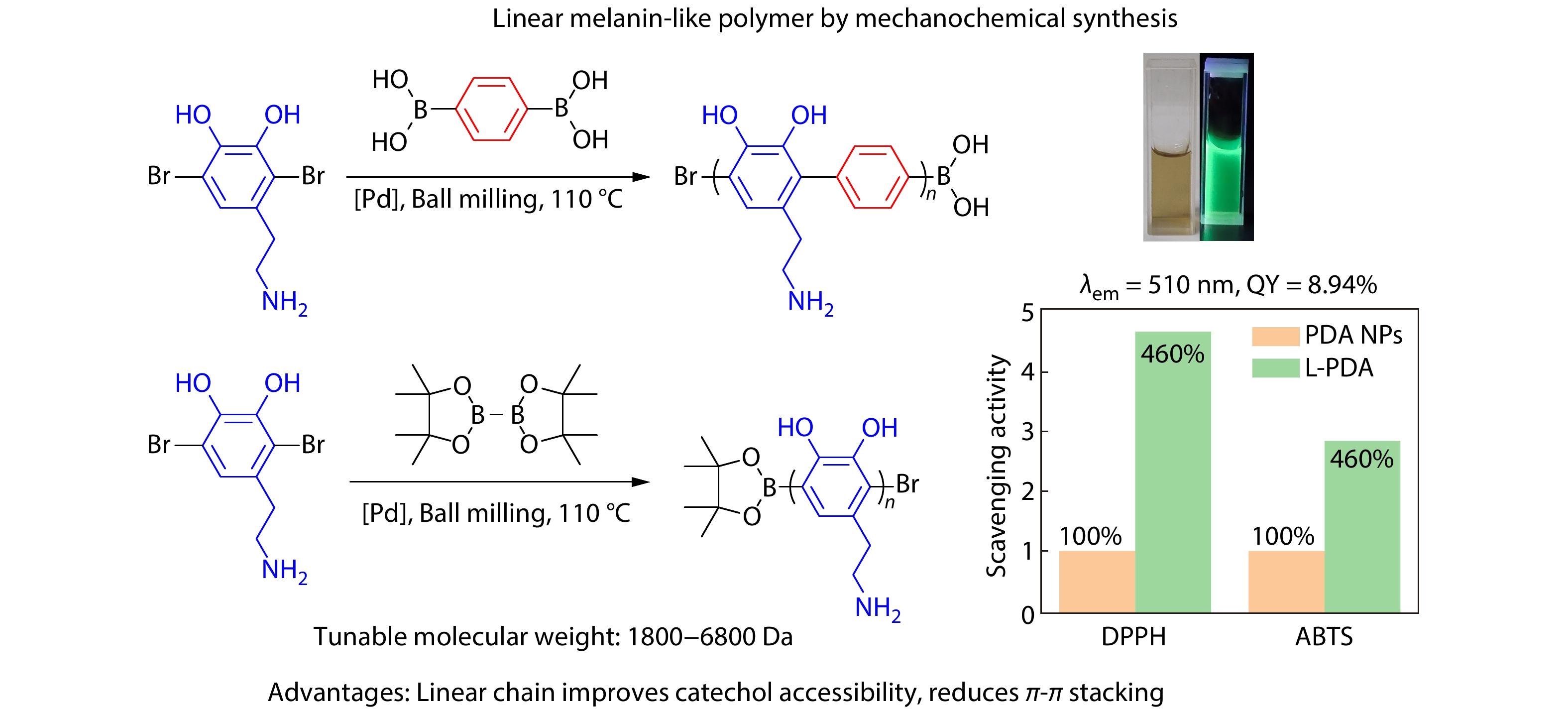
RESEARCH ARTICLE
2025, 43(6): 1043-1049. DOI: 10.1007/s10118-025-3313-yPublished(online): 2025-07-14Abstract Full text L-PDFAbstract
RESEARCH ARTICLE
2025, 43(6): 1050-1058. DOI: 10.1007/s10118-025-3328-4Published(online): 2025-07-14Abstract Full text L-PDFAbstract
RESEARCH ARTICLE
2025, 43(6): 1059-1066. DOI: 10.1007/s10118-025-3329-3Published(online): 2025-07-14Abstract Full text L-PDFAbstract
- Technical support is provided by Beijing Founder electronics co., LTD 京ICP备09064830号-19
 京公网安备11010802024621
京公网安备11010802024621 - It is recommended to read the content of this site in Chrome&IE9+. Please switch to extreme mode in browser 360.
- Cookies We use cookies to help provide and enhance our service and tailor content. By continuing, you agree to the use of cookies.
0