Issue Ebook
cover story

Latest Issue
Just Accepted Online First List of Issues Document Types
Issue 10, 2025
PREFACE
2025, 43(10): 1699. DOI: 10.1007/s10118-025-3442-3Published(online): 2025-09-26Full text L-PDF

RAPID COMMUNICATION
2025, 43(10): 1700-1706. DOI: 10.1007/s10118-025-3404-9Published(online): 2025-09-26Abstract Full text L-PDFAbstract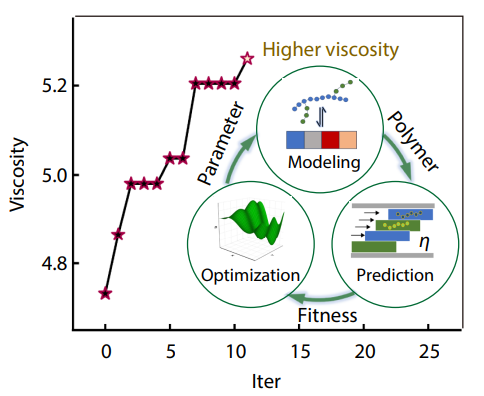
REVIEW
2025, 43(10): 1707-1717. DOI: 10.1007/s10118-025-3401-zPublished(online): 2025-09-26Abstract Full text L-PDFAbstract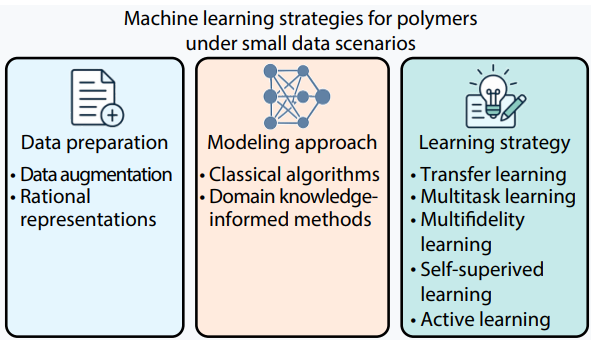
RESEARCH ARTICLE
2025, 43(10): 1718-1729. DOI: 10.1007/s10118-025-3403-xPublished(online): 2025-09-26Abstract Full text L-PDFAbstract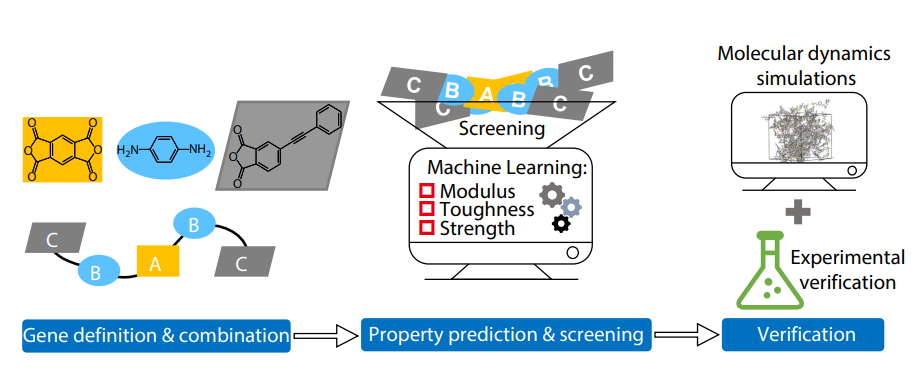
RESEARCH ARTICLE
2025, 43(10): 1730-1738. DOI: 10.1007/s10118-025-3396-5Published(online): 2025-09-26Abstract Full text L-PDFAbstract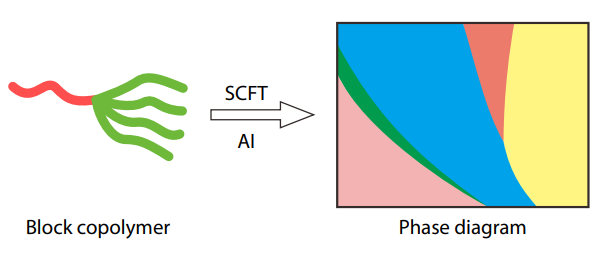
RESEARCH ARTICLE
2025, 43(10): 1739-1748. DOI: 10.1007/s10118-025-3380-0Published(online): 2025-09-26Abstract Full text L-PDFAbstract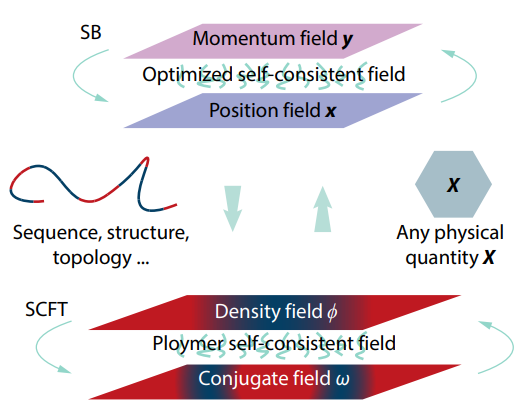
RESEARCH ARTICLE
2025, 43(10): 1749-1760. DOI: 10.1007/s10118-025-3402-yPublished(online): 2025-09-26Abstract Full text L-PDFAbstract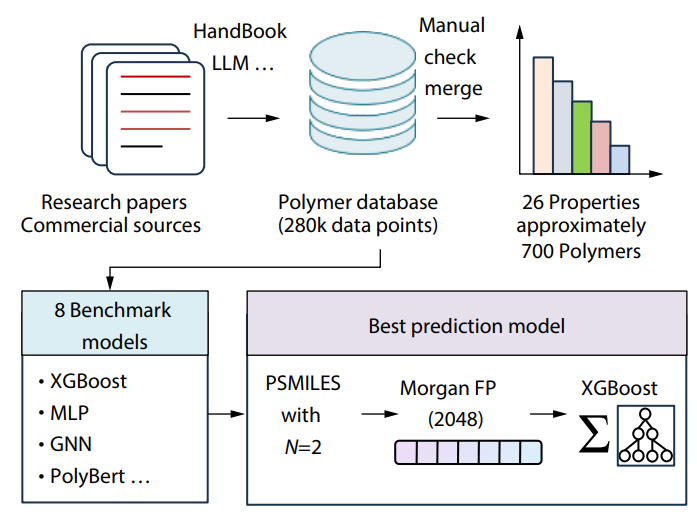
RESEARCH ARTICLE
2025, 43(10): 1761-1773. DOI: 10.1007/s10118-025-3389-4Published(online): 2025-09-26Abstract Full text L-PDFAbstract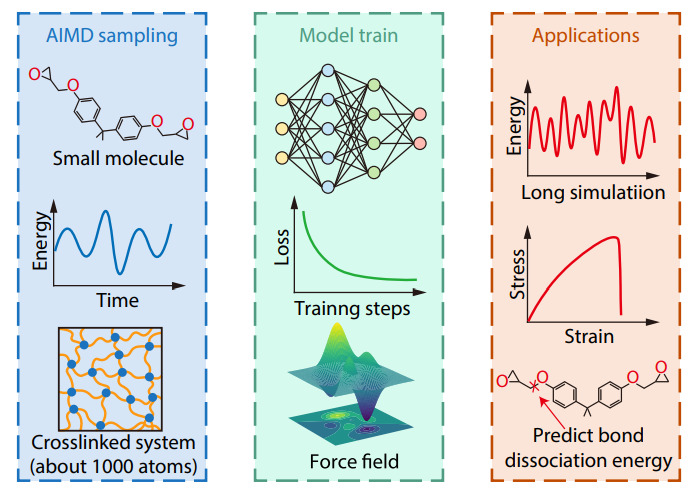
RESEARCH ARTICLE
2025, 43(10): 1774-1784. DOI: 10.1007/s10118-025-3392-9Published(online): 2025-09-26Abstract Full text L-PDFAbstract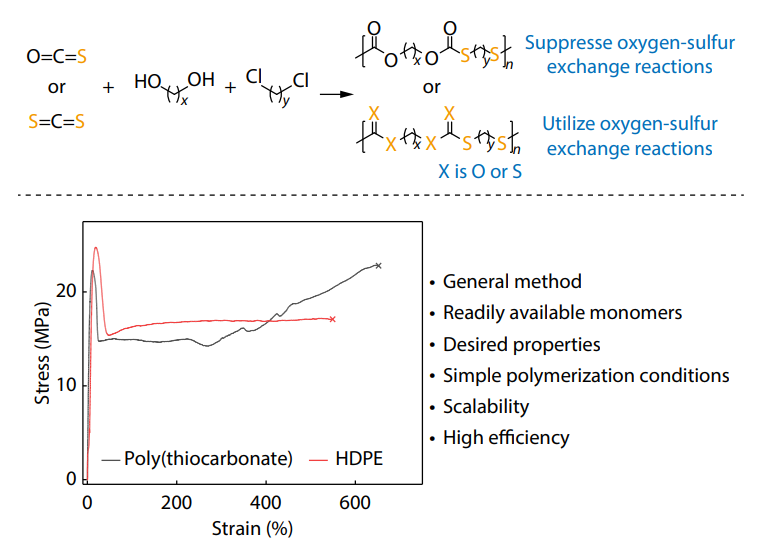
RESEARCH ARTICLE
2025, 43(10): 1785-1791. DOI: 10.1007/s10118-025-3393-8Published(online): 2025-09-26Abstract Full text L-PDFAbstract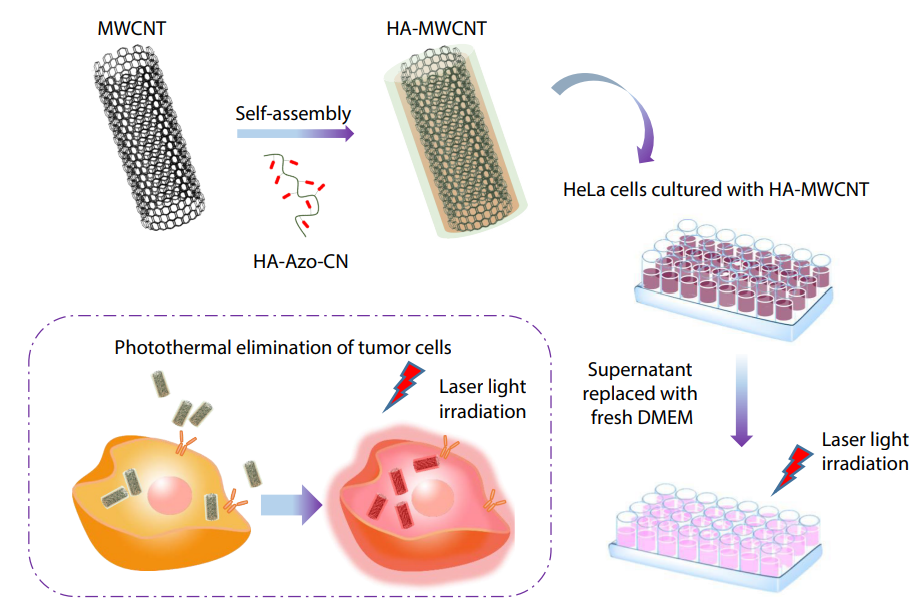
RESEARCH ARTICLE
2025, 43(10): 1792-1803. DOI: 10.1007/s10118-025-3395-6Published(online): 2025-09-26Abstract Full text L-PDFAbstract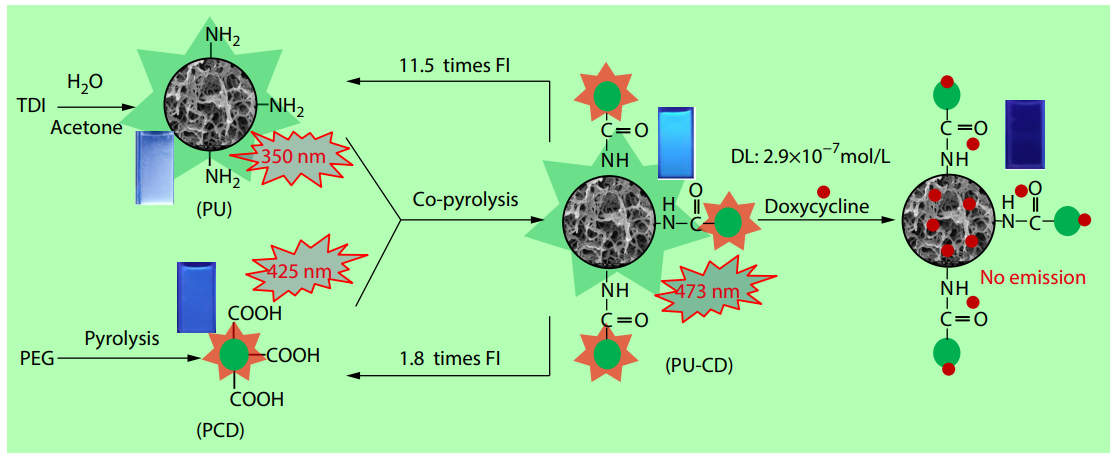
RESEARCH ARTICLE
2025, 43(10): 1804-1813. DOI: 10.1007/s10118-025-3415-6Published(online): 2025-09-26Abstract Full text L-PDFAbstract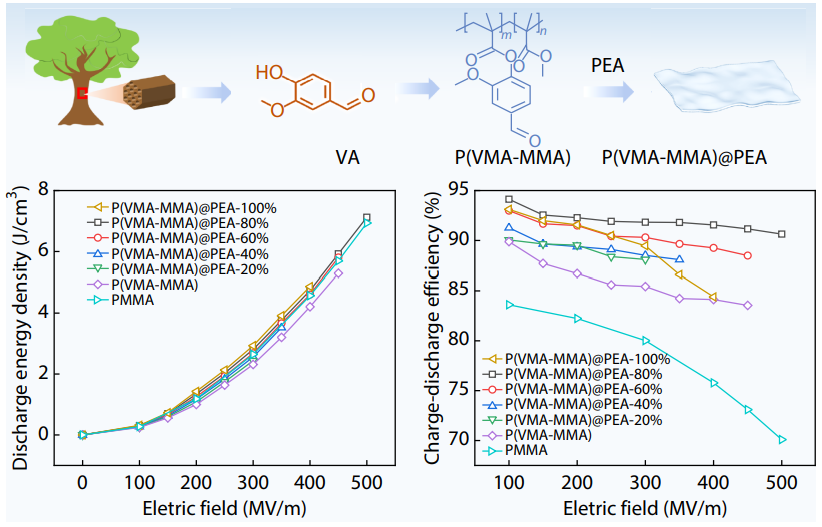
RESEARCH ARTICLE
2025, 43(10): 1814-1824. DOI: 10.1007/s10118-025-3397-4Published(online): 2025-09-26Abstract Full text L-PDFAbstract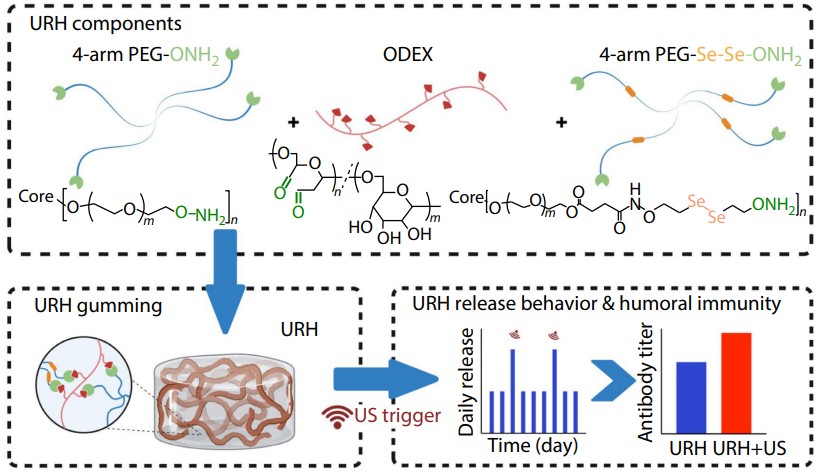
RESEARCH ARTICLE
2025, 43(10): 1825-1836. DOI: 10.1007/s10118-025-3408-5Published(online): 2025-09-26Abstract Full text L-PDFAbstract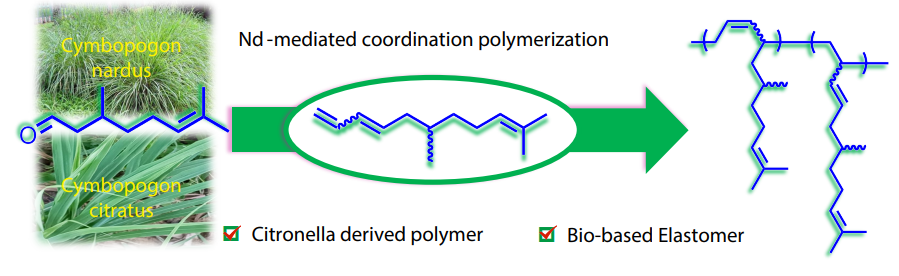
RESEARCH ARTICLE
2025, 43(10): 1837-1849. DOI: 10.1007/s10118-025-3409-4Published(online): 2025-09-26Abstract Full text L-PDFAbstract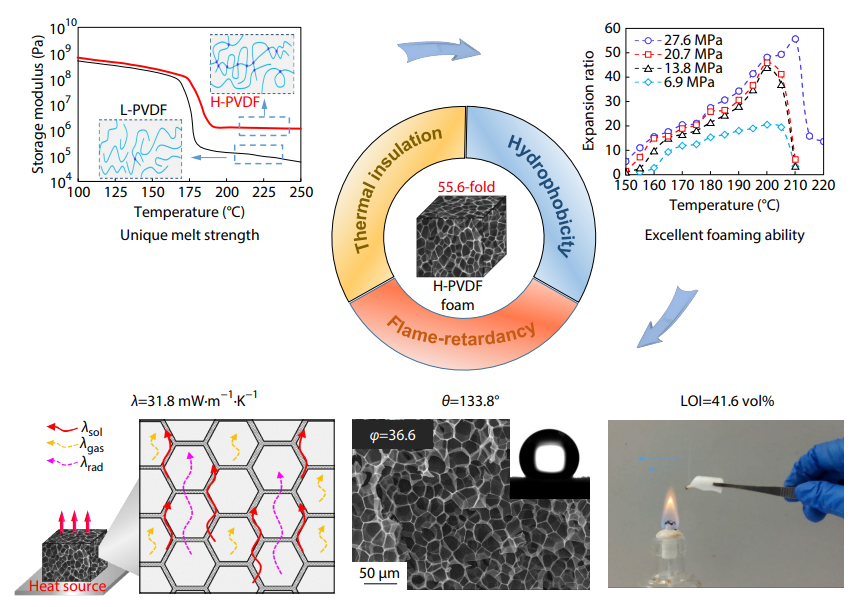
RESEARCH ARTICLE
2025, 43(10): 1850-1862. DOI: 10.1007/s10118-025-3390-yPublished(online): 2025-09-26Abstract Full text L-PDFAbstract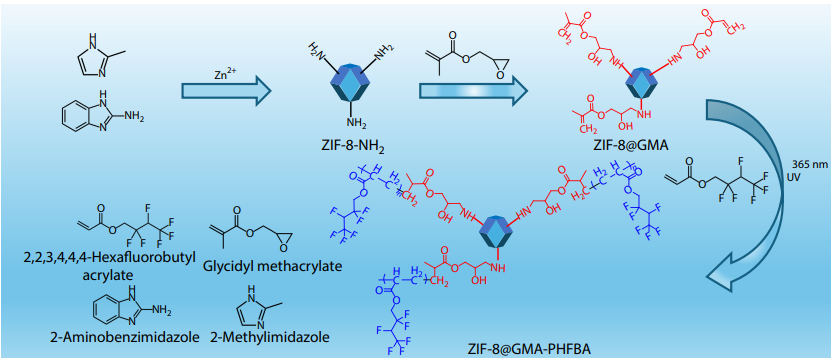
RESEARCH ARTICLE
2025, 43(10): 1863-1874. DOI: 10.1007/s10118-025-3410-yPublished(online): 2025-09-26Abstract Full text L-PDFAbstract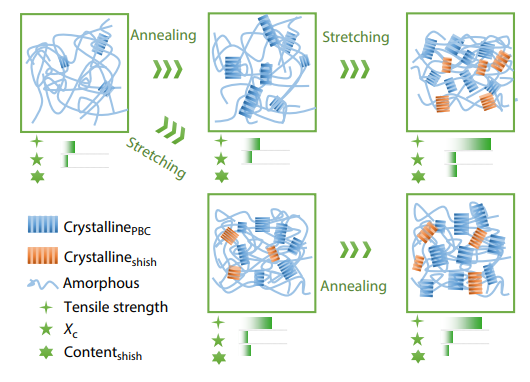
RESEARCH ARTICLE
2025, 43(10): 1875-1884. DOI: 10.1007/s10118-025-3384-9Published(online): 2025-09-26Abstract Full text L-PDFAbstract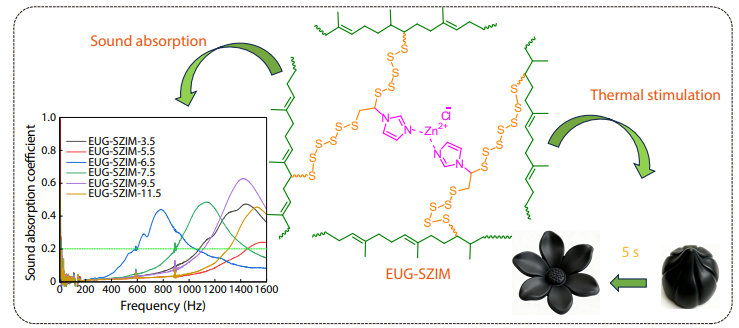
RESEARCH ARTICLE
2025, 43(10): 1885-1893. DOI: 10.1007/s10118-025-3386-7Published(online): 2025-09-26Abstract Full text L-PDFAbstract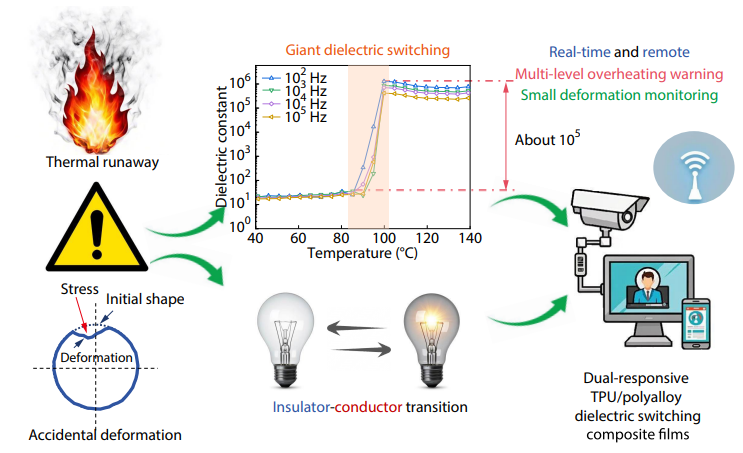
RESEARCH ARTICLE
2025, 43(10): 1894-1903. DOI: 10.1007/s10118-025-3406-7Published(online): 2025-09-26Abstract Full text L-PDFAbstract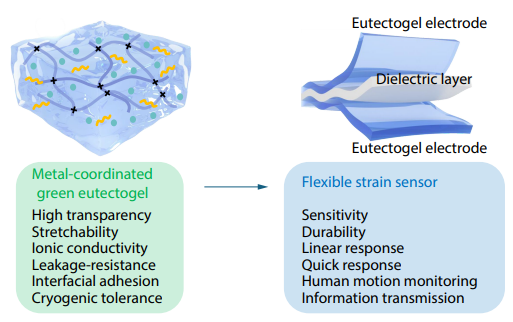
RESEARCH ARTICLE
2025, 43(10): 1904-1916. DOI: 10.1007/s10118-025-3407-6Published(online): 2025-09-26Abstract Full text L-PDFAbstract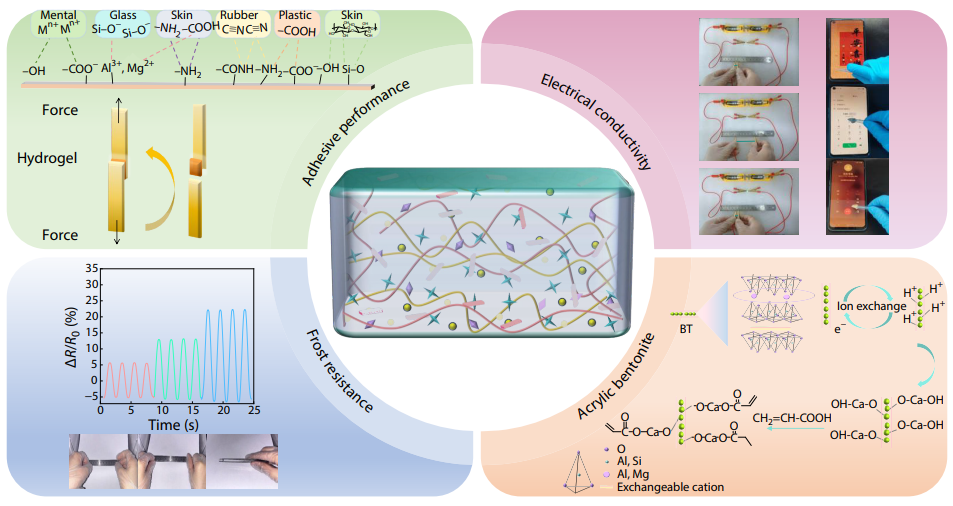
RESEARCH ARTICLE
2025, 43(10): 1917-1928. DOI: 10.1007/s10118-025-3391-xPublished(online): 2025-09-26Abstract Full text L-PDFAbstract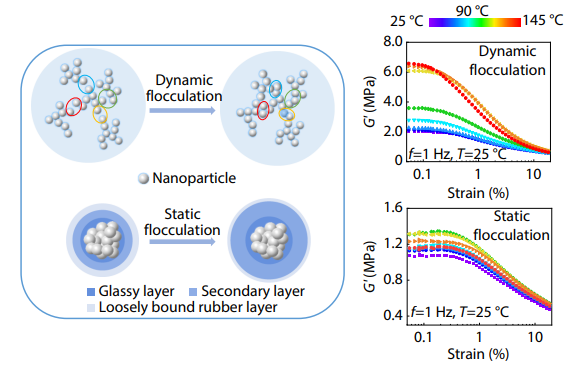
RESEARCH ARTICLE
2025, 43(10): 1929-1938. DOI: 10.1007/s10118-025-3400-0Published(online): 2025-09-26Abstract Full text L-PDFAbstract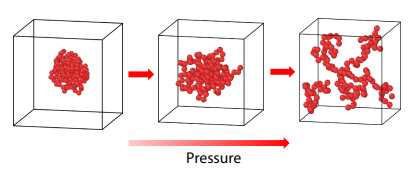
- Technical support is provided by Beijing Founder electronics co., LTD 京ICP备09064830号-19
 京公网安备11010802024621
京公网安备11010802024621 - It is recommended to read the content of this site in Chrome&IE9+. Please switch to extreme mode in browser 360.
- Cookies We use cookies to help provide and enhance our service and tailor content. By continuing, you agree to the use of cookies.
0